 The ATR-WEE1 kinase module inhibits the MAC complex to regulate replication stress response
The ATR-WEE1 kinase module inhibits the MAC complex to regulate replication stress response
Nucleic Acids Research


Lead contact.

- Altmetric
DNA damage response is a fundamental mechanism to maintain genome stability. The ATR-WEE1 kinase module plays a central role in response to replication stress. Although the ATR-WEE1 pathway has been well studied in yeasts and animals, how ATR-WEE1 functions in plants remains unclear. Through a genetic screen for suppressors of the Arabidopsis atr mutant, we found that loss of function of PRL1, a core subunit of the evolutionarily conserved MAC complex involved in alternative splicing, suppresses the hypersensitivity of atr and wee1 to replication stress. Biochemical studies revealed that WEE1 directly interacts with and phosphorylates PRL1 at Serine 145, which promotes PRL1 ubiquitination and subsequent degradation. In line with the genetic and biochemical data, replication stress induces intron retention of cell cycle genes including CYCD1;1 and CYCD3;1, which is abolished in wee1 but restored in wee1 prl1. Remarkably, co-expressing the coding sequences of CYCD1;1 and CYCD3;1 partially restores the root length and HU response in wee1 prl1. These data suggested that the ATR-WEE1 module inhibits the MAC complex to regulate replication stress responses. Our study discovered PRL1 or the MAC complex as a key downstream regulator of the ATR-WEE1 module and revealed a novel cell cycle control mechanism.
INTRODUCTION
DNA is constantly damaged by exogenous and endogenous factors, leading to various types of DNA lesions such as double-strand breaks (DSB), single-strand DNA (ssDNA) breaks, and crosslinks. To maintain genome integrity, all organisms have evolved elaborate and efficient DNA damage response (DDR) mechanisms, including activation of cell cycle checkpoints, DNA repair, transcriptional reprogramming, and apoptosis. The two evolutionarily conserved protein kinases ATM (ataxia telangiectasia mutated) and ATR (ATM and RAD3-related) are the master regulators of DDR. In general, ATM is activated by DSB, whereas ATR mainly responds to ssDNA and stalled replication forks. Defects in DDR are the causes of many diseases including cancers (1–3).
In animals, the ATR pathway has been well-studied (4,5). When activated by replication stress, ATR phosphorylates checkpoint kinase 1 (CHK1), which in turn activates WEE1 kinase and inhibits CDC25 phosphatase. While WEE1 inhibits cyclin-dependent kinases (CDKs), the key drivers of cell cycle progression, by phosphorylating the conserved Thr14 and Tyr15, CDC25 activates CDKs by dephosphorylating the same residues (6–10). The Arabidopsis genome encodes both ATR and WEE1 homologs, but lack functional CHK1 or CDC25 homologs (11). In addition, the CDKA;1 containing substitutions of Thr14 and Tyr15 with nonphosphorylatable Val and Phe could fully complement the cdka;1 mutant under both normal and replication stress conditions (11). Based on this result, it was proposed that WEE1 activates cell cycle arrest independently of the phosphorylation of CDKA;1 (11). Therefore, how ATR and WEE1 regulate cell cycle checkpoints in plants is still unknown. It is possible that WEE1 may function by phosphorylating other substrates or other residues of CDKA;1.
The PRP19 complex (Prp19C), also known as the NineTeen Complex (NTC) in yeasts and the MOS4-associated complex (MAC) in plants, is evolutionarily conserved. In humans, the core components of this complex include PRP19, CDC5L, PLRG1 and SPF27, whose homologs in plants are MAC3, CDC5, PRL1 and MOS4, respectively. The Prp19C complex was originally discovered to function in RNA splicing (12–16). Accumulating evidence suggests that Prp19C plays important roles in DDR (17,18). Recently, two groups independently found that PRP19 is essential for ATR activation in response to replication stress (19,20). In Arabidopsis, the MAC complex is reported to control RNA splicing and miRNA biogenesis (16,21–23). Physiologically, it was shown that the MAC complex regulates stem cell maintenance and immunity (24–28). However, it is still unknown whether and how this complex regulates DDR in plants.
In animals, loss of function of ATR or WEE1 results in embryonic lethality (29,30). However, the Arabidopsis atr and wee1 mutants grow normally, indicating that ATR and WEE1 may function differently from their animal homologs. It was reported that both atr and wee1 are hypersensitive to replication-blocking agent hydroxyurea (HU), resulting in a short-root phenotype (31,32). Based on this phenotype, we performed a genetic screen for suppressors of atr (soat) to elucidate how ATR regulates replication stress response in plants. In this study, we characterized one of the suppressors, soat2. SOAT2 encodes PRL1 protein, which is a core subunit of the MAC complex in Arabidopsis. Further studies revealed that loss of function of PRL1 also suppresses the hypersensitivity of wee1 to HU. Mechanistically, WEE1 directly interacts with and phosphorylates PRL1 at Serine 145, which promotes PRL1 ubiquitination and subsequent degradation through 26S proteasome. In support of the roles of PRL1 in both RNA splicing and DDR, loss of function of PRL1 induces intron retention of cell cycle genes including CYCD1;1 and CYCD3;1, which is abolished in wee1 but restored in wee1 prl1. Co-expressing the coding sequences of CYCD1:1 and CYCD3;1 in wee1 prl1 partially restores the root length and HU sensitivity. Based on these results, we propose that the ATR-WEE1 module regulates replication stress response by inhibiting the MAC complex to induce intron retention of cell cycle genes. Our study not only discovered PRL1 or the MAC complex as a key downstream regulator of the ATR-WEE1 module but also revealed a novel cell cycle control mechanism.
MATERIALS AND METHODS
Materials and growth conditions
All Arabidopsis thaliana mutants used in this study are in the Columbia (Col-0) background. The atr (SALK_032841), wee1 (SALK_147968C) and cdc5 mutant (CS426613) mutants were obtained from ABRC. The mcr1 mutant was described previously (28). Seeds were sterilized with 2% PPM (Plant Cell Technology), stratified at 4°C in the dark for 2 days, and then plated on 1/2 Murashige and Skoog (MS) medium containing 1% sucrose and 0.3% phytagel. The plants were grown under long-day conditions (16 h of light and 8 h of dark) at 22°C in a growth chamber. The calluses were induced on MS medium containing 0.5 mg/l 2,4-D and 0.05 mg/l kinetin. The primers used in the study were listed in Supplementary Table S1.
Mutant screening
The activation-tagging vector pBASTA-AT2 (33) containing the herbicide selectable marker gene BASTA was transformed into the atr mutant using the Agrobacterium-mediated Arabidopsis floral-dip method (34). The T2 seeds were screened for suppressors of atr. The seedlings were grown on 1/2 MS media containing 0.75 mM HU, and the root length of plants was examined after 7–10 days. The plants with longer roots were considered to be the suppressors of atr.
Quantitative reverse transcription PCR (qRT-PCR)
Samples were collected and quickly frozen in liquid nitrogen and stored −80°C until use. Total RNA was extracted using TRIzol reagent (Invitrogen). The RNA integrity was examined by running the RNA samples on a 1% agarose gel. The HiFiScript gDNA Removal cDNA Synthesis Kit (CW2582, CoWin Biosciences) were used for gDNA removal and cDNA synthesis. To confirm that the genomic DNA was removed, the RNA samples were used as templates in PCR reactions using primer pairs flanking the intron regions of ACTIN7 or EMB2386. For cDNA synthesis, 1 μg RNA was used in a 20 μl reaction according to the manual's instructions. The cDNA samples were diluted five times and used as templates in qPCR using Ultra SYBR Mixture kit (CW0957M, CoWin Biosciences). The reactions were as follows: 10 μl of 2× qPCR mixture, 0.4 μl of 10 μM forward primer, 0.4 μl of 10 μM reverse primer, 4 μl cDNA, 5.2 μl PCR grade water. PCR was performed using Bio-Rad CFX 96 detection system with the following parameters: 95°C 3 min, 40 cycles of 95°C 15 s, 56°C 15 s and 72°C 30 s. To validate qPCR, we performed a standard curve using serials of samples with dilution factor of 1:5. For data analysis, relative transcript abundance was calculated by 2−△△CT method.
Generation of PRL1 antibody
The PRL1 antibody was generated by Atagenix Laboratories (Wuhan, China). The peptide VVSQPPRQPDRINEQPGPS was used as the antigen as described previously (24).
In vitro pull-down assay
MBP-PRL1, GST, and GST-WEE1 proteins were expressed in Escherichia coli BL21(DE3). MBP-PRL1 was purified using Amylose Resin (New England BioLabs). GST and GST-WEE1 were coupled to Glutathione beads (GE Healthcare Life Sciences) and then incubated with MBP-PRL1 protein in the binding buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA and 2 mM DTT) at 4°C for 2 h. The beads were washed three times with washing buffer (binding buffer plus 2% NP-40), boiled in 1× SDS loading buffer, and analyzed by western blot using anti-MBP antibody (ABclonal).
Co-immunoprecipitation assay
The 35S:GFP, 35S:PRL1-GFP and 35S:WEE1-FLAG were transformed into Agrobacterium tumefaciens GV3101. The 35S:WEE1-FLAG strain was co-infiltrated with 35S:GFP or 35S:PRL1-GFP into the leaves of N. benthamiana. After 48 h, the infiltrated leaves were ground in liquid nitrogen and were resuspended in IP buffer (20 mM Tris–HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 1 mM PMSF, 100 μM MG132, 1× protease inhibitor cocktail) for total protein extraction. The lysates were incubated with GFP-Trap magnetic beads (Chromotek) at 4°C for 2 h. The beads were washed using washing buffer (20 mM Tris–HCl pH 7.5, 150–500 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1× Protease Inhibitor Cocktail) and then boiled in 1× SDS loading buffer. The western blotting was performed using anti-FLAG (Promoter) and anti-GFP (Promoter) antibodies.
Split luciferase assay
Split luciferase assay was performed as described previously (35). The genes were cloned into the vectors containing either the C-terminal half of luciferase (CLuc) or the N-terminal half of luciferase (NLuc), and then the constructs were transformed into Agrobacterium tumefaciens strain GV3101, respectively. The resultant CLuc and NLuc strains were co-infiltrated into leaves of N. benthamiana. After 48 h, 1 mM luciferin was applied onto leaves and the images were captured using Lumazone imaging system equipped with 2048B CCD camera (Roper).
Immunoprecipitation of GFP proteins from calluses
The calluses derived from pPRL1:PRL1-GFP/Col-0 and pPRL1:PRL1-GFP/wee1 were treated with or without HU. After being ground in liquid nitrogen (0.2 g), they were resuspended in 400 μl IP buffer and incubated on ice for 20 min, followed by centrifugation at 20 000 × g at 4°C for 10 min. The supernatant was incubated with 25 μl GFP-Trap magnetic beads (Chromotek) at 4°C for 3 h. The beads were washed four times with washing buffer and subjected for subsequent assays.
In vitro phosphorylation assay
MBP-PRL1, MBP-prl1S145A, GST-WEE1, and GST-wee1kd were expressed in E. coli and purified. To test whether WEE1 phosphorylates PRL1, MBP-PRL1 or MBP-prl1S145A was incubated with GST-WEE1 or GST-wee1kd in wee1 extracts containing 25 mM Tris–HCl pH 7.5, 10 mM NaCl, 10 mM MgCl2, 4 mM PMSF, 5 mM DTT, 10 mM ATP at 37°C for 30 min. To test whether HU treatment affected PRL1 phosphorylation level, MBP-PRL1 was incubated with extracts of Col-0 and wee1 treated with or without HU at 37°C for 30 min. The reactions were stopped by adding 2× SDS loading buffer followed by western blotting analysis. The phosphorylation levels were determined using an anti-phosphoserine/threonine antibody (ECM Biosciences).
In vitro ubiquitination assay
The in vitro ubiquitination assay was performed as described previously with some modifications (36). The MBP-PRL1 protein was incubated with Amylose Resin (NEB) at 4°C for 2 h, and the beads were washed three times with PBS buffer. Total proteins were extracted from calluses with native protein extraction buffer (25 mM Tris–HCl PH 7.5, 10 mM NaCl, 10 mM MgCl2, 4 mM PMSF, 5 mM DTT, 10 mM ATP, 100 μM MG132). Equal amounts of beads were incubated with equal amounts of Col-0 or wee1 extracts at room temperature for 4 h. The beads were then washed four times and boiled in 1× SDS loading buffer, followed by western blotting analysis using anti-ubiquitin antibody (CST).
In vitro degradation assay
The in vitro degradation assay was performed as described previously (37). Total proteins were extracted from calluses with native protein extraction buffer (25 mM Tris–HCl pH 7.5, 10 mM NaCl, 10 mM MgCl2, 4 mM PMSF, 5 mM DTT, 10 mM ATP). Equal amounts of proteins from Col-0 and wee1 mutant were incubated with MBP-PRL1 recombinant protein at 22°C for different times (0, 2, 4, 8 h). PRL1 protein was detected by western blotting using anti-MBP antibody (ABclonal).
In vivo degradation assay
The Col-0 and wee1 calluses were treated with 100 μM cycloheximide (CHX) for different times (0, 2, 4, 8, 12 h) to block protein biosynthesis. Total proteins were extracted using RIPA buffer and subjected to western blotting analysis using anti-PRL1 antibody.
Transient expression in protoplasts
Isolation of Arabidopsis mesophyll protoplasts and PEG-induced transfection were performed as described previously (38), with some modifications. Briefly, the leaves from 3-week-old plants grown in a short photoperiod greenhouse (12 h light and 12 h dark at 22°C) were cut into 0.5–1 mm leaf strips and digested in the enzyme solution containing 1.5% cellulase R10 and 0.5% macerozyme for 3–4 h. The protoplasts were isolated from the enzyme solution by filtration through two-layers Miracloth (Millipore). Plasmids were transfected into protoplasts by PEG-calcium methods. After culturing for 12 h, the protoplasts were harvested and subjected for western blotting analysis.
Mass spectrometry
The PRL1-GFP proteins were immunoprecipitated using GFP-Trap from the pPRL1: PRL1-GFP/Col-0 treated with or without 10 mM HU. The immunoprecipitated proteins were digested on the beads with trypsin at 37°C overnight, and the resultant peptides were analyzed on an Ultimate 3000 nano UHPLC system (Thermo Scientific) coupled online to a hybrid Quadrupole-Orbitrap mass spectrometer Q Exactive HF (Thermo Scientific). The raw file was processed using Peaks Studio version 8.5 (Bioinformatics Solutions) by Peaks search engine with the Araport11 protein database (total 48 359 entries).
Co-expression of CYCD3;1 and CYCD1;1
CYCD3;1 and CYCD1;1 were co-expressed in Arabidopsis using IntF2A (Intein-linker-F2A)-based polyprotein cassette (39). The coding sequence of CYCD3;1 fused with 3 × FLAG, the IntF2A domain, and the coding sequence of CYCD1;1 fused with GFP were cloned into pFGC5941. The resultant vector was transformed into Col-0 and wee1 mcr1 by Agrobacterium-mediated floral dipping method (34).
Statistical analysis
Statistical tests were performed using GraphPad Prism 5.
RESULTS
Identification of the atr suppressor soat2
To study how ATR functions in Arabidopsis, we performed a genetic screen for suppressors of atr using an activation-tagging strategy, aiming to identify both negative and positive regulators in the ATR pathway. The activation-tagging vector pBASTA-AT2 (33) was transformed into the atr mutant using the floral-dip method (34). The resulting T2 plants were grown vertically on medium containing 0.75 mM HU to screen for plants with roots longer than atr. One of the suppressors identified was soat2. As shown in Figure 1A and B, the root length of atr mutant was dramatically reduced when treated with HU. Although the roots of atr soat2 were much shorter than wild-type Col-0 and atr in the absence of HU, they were longer than atr when treated with HU. Statistical analysis indicated that the sensitivity of atr soat2 to HU was reduced significantly compared to atr. To exclude the possibility that the soat2 mutation rendered plants insensitive to any stimuli, we treated these plants with another DNA-damaging agent bleomycin (BLM), which causes DSBs. Unlike the HU response, the root length of atr soat2 mutant was dramatically reduced by BLM treatment (Figure 1C and D). Statistical analysis indicated that the atr soat2 mutant was significantly more sensitive to BLM than the atr mutant. These data indicated that the soat2 mutation specifically suppressed the hypersensitivity of atr to HU.

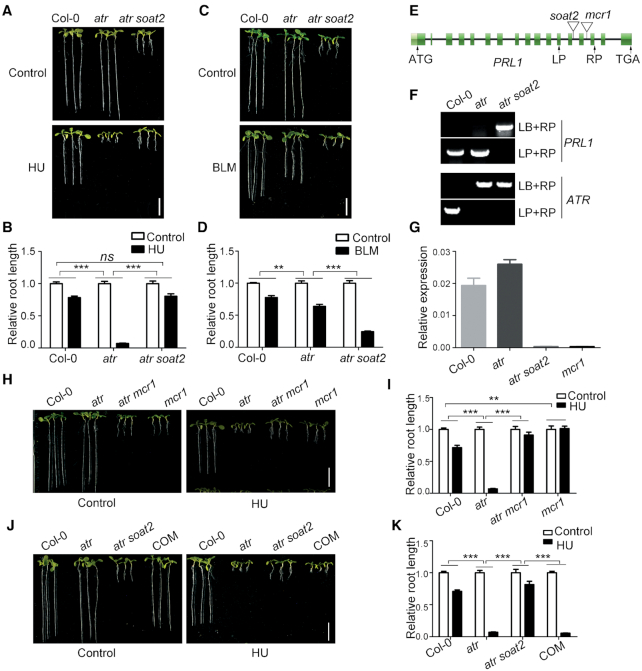
Loss of function of PRL1 suppresses atr mutant. (A, C, H and J) Pictures of plants treated with hydroxyurea (HU, a replication inhibitor) or bleomycin (BLM, a chemical inducing DNA double-strand breaks). The plants were grown vertically on 1/2 MS medium with or without 0.75 mM HU or 2.5 μM BLM for 8 days. COM, the complementation line in which the genomic PRL1 driven by its native promoter (pPRL1:PRL1) were transformed into atr soat2. Bar = 1 cm. (B, D, I and K) The relative root length of plants treated with HU or BLM. The relative root length data are represented as means ± SD (n = 20) relative to the values obtained under the control condition. (E) The genomic structure of PRL1. The exons are shown as green boxes, and introns are represented by black lines. ATG and TGA indicate the start and stop codons, respectively. The T-DNA insertion sites of soat2 and mcr1 are shown. (F) Genotype identification of PRL1 and ATR in atr and atr soat2. LP and RP are primers flanking the insertion sites. LB is the primer on the left border of T-DNA. (G) Relative expression of PRL1 determined by qRT-PCR analysis. The UBQ5 was used as a reference gene. Data represent mean ± SD (n = 3). The statistical significance was determined using two-way ANOVA analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significance. All experiments were repeated three times with similar results.
SOAT2 encodes PRL1
To determine the T-DNA insertion site in soat2, we carried out inverse PCR analysis followed by DNA sequencing (Supplementary Figure S1). We found that the T-DNA was inserted into the thirteenth intron of the PRL1 gene (Figure 1E). Genotype identification further confirmed that the T-DNA insertion was homozygous in atr soat2 (Figure 1F). We also tested the transcript level of PRL1 in Col-0, atr, and atr soat2 through qRT-PCR. Compared to Col-0, the transcript of PRL1 in atr soat2 was negligible, indicating that soat2 is a knockout mutant of PRL1 (Figure 1G).
To confirm that the PRL1 mutation suppresses atr, we crossed atr with mcr1, another T-DNA insertion mutant of PRL1 (28). Similar to atr soat2, the resulting atr mcr1 double mutant also suppressed the hypersensitivity of atr to HU (Figure 1H and I). In addition, we found that the mcr1 mutant was less sensitive to HU compared to Col-0. To further confirm our results, we transformed the genomic PRL1 driven by the native PRL1 promoter (pPRL1:PRL1) into atr soat2. The resulting complementation lines (COM) displayed the same phenotypes as the atr mutant, indicating that PRL1 could complement the soat2 mutant (Figure 1J and K).
Loss of function of CDC5 also suppresses the atr mutant
CDC5 is another core component of the MAC complex (25,40,41). To test whether suppression of atr is attributed to the specific function of PRL1 or the general function of the MAC complex, we crossed atr with cdc5 mutant to obtain the atr cdc5 double mutant and tested their response to HU treatment. Similar to atr mcr1, the roots of atr cdc5 were much shorter than atr in the absence of HU but were longer than atr when treated with HU (Figure 2A and B), indicating that loss of function of CDC5 can also suppress the atr mutant. As expected, similar to the atr mcr1 mutant, the atr cdc5 mutant was more sensitive to BLM than atr. These results suggest that loss of function of the MAC complex suppresses the hypersensitivity of atr to replication stress.
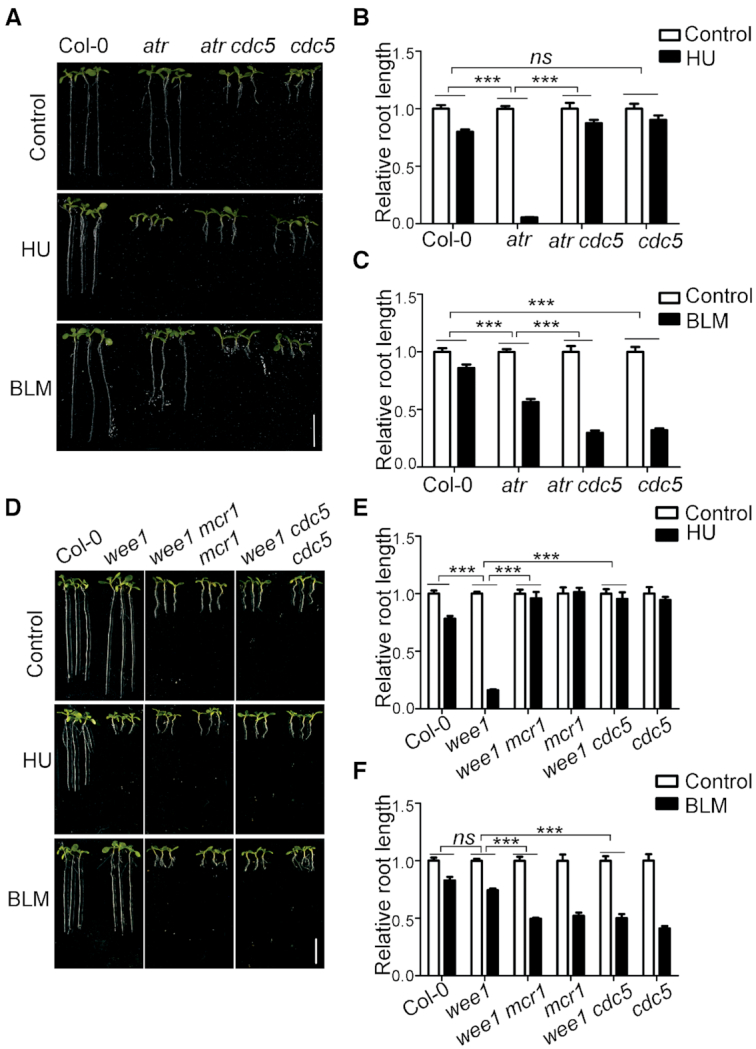
Loss of functions of PRL1 and CDC5 suppress wee1 mutant. (A and D) Pictures of plants treated with HU or BLM. The plants were grown vertically on 1/2 MS medium with or without 0.75 mM HU or 2.5 μM BLM for 8 days. Bar = 1 cm. (B, C, E and F) The relative root length of plants treated with HU or BLM. The relative root length data are represented as means ± SD (n = 20) relative to the values obtained under the control condition. The statistical significance was determined using Two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significance. All experiments were repeated three times with similar results.
Loss of functions of PRL1 and CDC5 suppress the wee1 mutant
The suppression of the hypersensitivity of atr to HU by loss of functions of PRL1 and CDC5 suggests that ATR negatively regulates PRL1 and CDC5 possibly through interaction and phosphorylation. However, we failed to detect the interactions between ATR and PRL1 or CDC5, indicating that ATR may indirectly regulate PRL1 and CDC5. Previous studies suggested ATR induces WEE1 and loss of function of WEE1 leads to plant hypersensitivity to HU (32). Therefore, it is likely that ATR regulates PRL1 and CDC5 through its effects on WEE1. We proposed that loss of function of PRL1 or CDC5 should also suppress the hypersensitivity of wee1 to HU. To test this hypothesis, we generated the wee1 mcr1 and wee1 cdc5 double mutants by crossing wee1 with mcr1 or cdc5. As shown in Figure 2D and E, compared to Col-0, the wee1 mutant was hypersensitive to HU. Although the roots of wee1 mcr1 and wee1 cdc5 mutants were much shorter than wee1 in the absence of HU, they were even longer than wee1 in the presence of HU, indicating that wee1 mcr1 and wee1 cdc5 were less sensitive to HU than wee1. In contrast, wee1 mcr1 and wee1 cdc5 were more sensitive to BLM than wee1 (Figure 2E and F). These results suggested that mcr1 and cdc5 specifically suppress the hypersensitivity of wee1 to replication stress, indicating that WEE1 negatively regulates PRL1 and CDC5.
WEE1 interacts with PRL1
Given that WEE1 is a protein kinase, we propose that WEE1 may interact with and phosphorylate PRL1. To test this hypothesis, we first carried out pull-down assays to investigate their interaction. The glutathione S-transferase (GST)-tagged WEE1 (GST-WEE1) and maltose-binding-protein (MBP)-tagged PRL1 (MBP-PRL1) were expressed in E. coli. As shown in Figure 3A, GST-WEE1, but not GST, could pull down MBP-PRL1 protein, indicating that WEE1 directly interacts with PRL1. To test whether they can interact in vivo, we performed coimmunoprecipitation (Co-IP) assays. The WEE1-FLAG fusion protein was co-expressed with PRL1-GFP or GFP in N. benthamiana. The proteins were immunoprecipitated using GFP-Trap and subjected to western blotting analysis. In line with the pull-down results, WEE1-FLAG protein could be coimmunoprecipitated by PRL1-GFP, but not the GFP control (Figure 3B). To further confirm their in vivo interaction, we conducted split luciferase assays in N. benthamiana (42). PRL1 was fused with the C-terminal half of luciferase (CLuc) and WEE1 was fused with the N-terminal half of luciferase (NLuc). An interaction between two proteins brings the two halves of the luciferase together, leading to enzymatic activity and production of luminescence that is detectable using a hypersensitive CCD camera. As shown in Figure 3C, the luminescence signal could be detected only when cLuc-PRL1 and WEE1-nLuc were co-expressed, indicating that WEE1 interacts with PRL1 in vivo.
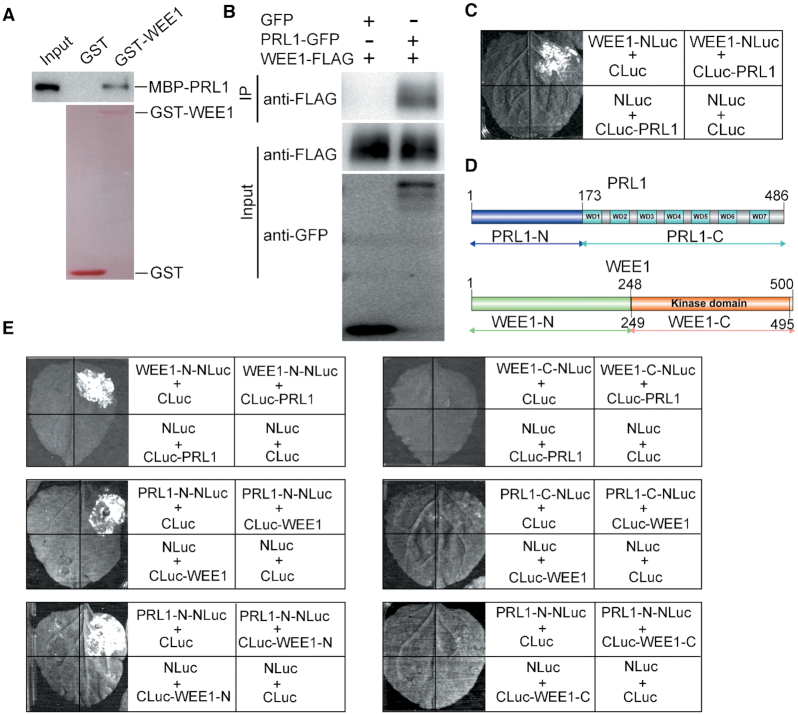
PRL1 interacts with WEE1. (A) In vitro pull-down assays. The recombinant GST or GST-WEE1 proteins were coupled to glutathione beads and incubated with the recombinant MBP-PRL1 proteins. MBP-PRL1 was pulled down by GST-WEE1, but not GST. The GST and GST-WEE1 proteins were detected through Ponceau S staining. MBP-PRL1 was detected using anti-MBP antibody. (B) Co-immunoprecipitation assays. WEE1-FLAG was co-expressed with PRL1-GFP or GFP in N. benthamiana leaves. Immunoprecipitation was performed using GFP-Trap beads and western blotting was performed using anti-FLAG or anti-GFP antibodies. (C, E) Split luciferase assays. The Agrobacterium bacteria carrying the indicated constructs were co-expressed in N. benthamiana leaves. The positive luminescence detected by a CCD camera indicates interaction. (D) Outline of PRL1 and WEE1 structures, highlighting the conserved domains. The truncation sites of PRL1 and WEE1 were indicated. All experiments were repeated three times with similar results.
To study how WEE1 regulates PRL1, we sought to map the interacting domain between WEE1 and PRL1. WEE1 contains a kinase domain at the C-terminus and PRL1 contains 7 WD40 motifs at the C-terminus (Figure 3D). Both WEE1 and PRL1 were truncated into N-terminal and C-terminal halves according to their domain structure (Figure 3D). Split luciferase assays were used to test their interactions. As shown in Figure 3E, WEE1-N could interact with the full-length PRL1 and PRL1-N could interact with the full-length WEE1. Furthermore, we found that PRL1-N could interact with WEE1-N. These results indicate that the N-terminus of PRL1 is responsible for interaction with WEE1, and the C-terminus of PRL1 may mediate its interactions with other proteins. Since our data showed that the N-terminus of WEE1 is responsible for interaction with PRL1, we proposed that the kinase-domain-containing C-terminus may function to phosphorylate PRL1.
WEE1 phosphorylates PRL1 at serine 145
Next, we investigated whether WEE1 can phosphorylate PRL1. To this end, we performed in vitro phosphorylation assays. We substituted the conserved Asp 372 in the catalytic domain of WEE1 to Asn to make a kinase-dead form of WEE1 (wee1kd), which was used as a strict negative control. The recombinant MBP-PRL1, GST-WEE1, or GST-wee1kd were incubated with the wee1 extracts and subjected to western blotting using an antibody that recognizes phosphorylated serine and threonine residues (anti-pS/pT). As shown in Figure 4A, MBP-PRL1 incubated with wee1 extracts was weakly phosphorylated, indicating that PRL1 may be phosphorylated by other kinases in addition to WEE1. The phosphorylation was dramatically enhanced when GST-WEE1 was added into the reaction. However, the kinase-dead GST-wee1kd could not enhance the phosphorylation. These results indicate that WEE1 promotes PRL1 phosphorylation.
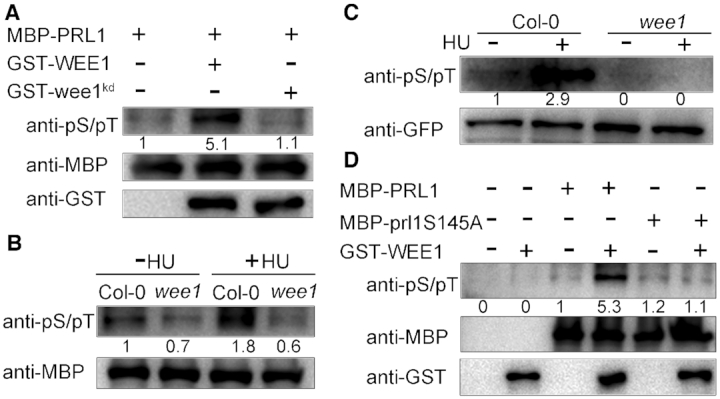
WEE1 phosphorylates PRL1. (A, B and D) In vitro phosphorylation assays. MBP-PRL1, MBP-prl1S145A, GST-WEE1 and GST-wee1kd were expressed in E. coli and purified. wee1kd indicates kinase-dead form of WEE1. prl1S145A indicates the non-phosphorylatable PRL1 with replacement of Ser145 by Ala. The indicated proteins were incubated in extracts of wee1 (A and D). MBP-PRL1 proteins were incubated with extracts of Col-0 or wee1 treated with (+) or without (−) HU (B). Phosphorylation levels were determined using an antibody that recognizes phosphorylated serine and threonine residues (anti-pS/pT). (C) In vivo phosphorylation assays. The PRL1-GFP proteins were immunoprecipitated using GFP-Trap from the transgenic PRL1-GFP plants in Col-0 and wee1 background treated with (+) or without (−) 1 mM HU and then subjected to western blotting using anti-pS/pT and anti-GFP antibodies. The experiments were repeated three times with similar results.
To confirm that WEE1 promotes PRL1 phosphorylation and to test whether PRL1 phosphorylation is further enhanced by HU treatment, we compared the phosphorylation level of MBP-PRL1 using the extracts of Col-0 and wee1 treated with or without HU. As shown in Figure 4B, the MBP-PRL1 phosphorylation level was higher in Col-0 than in wee1. In addition, HU treatment could significantly enhance the phosphorylation level of MBP-PRL1 in Col-0, but not in the wee1 mutant.
To further confirm that WEE1 phosphorylates PRL1, we examined the phosphorylation level of PRL1 in planta. We generated transgenic plants containing GFP-tagged PRL1 driven by its native promoter (pPRL1:PRL1-GFP) in Col-0 and wee1 backgrounds. The calluses derived from the transgenic plants were treated with or without HU. The PRL1-GFP proteins were immunoprecipitated using GFP-Trap and subjected to western blotting analysis. The phosphorylation level of PRL1 in Col-0 was much stronger than in wee1 (Figure 4C). In addition, we found that HU treatment could enhance PRL1 phosphorylation in Col-0, but not in the wee1 mutant. Taken together, these results suggested that WEE1 phosphorylates PRL1 and HU treatment enhances this phosphorylation.
To identify the phosphorylation sites in PRL1, we performed mass spectrometry analysis using PRL1-GFP protein immunoprecipitated from calluses treated with or without HU. We found that Ser145 was phosphorylated and this phosphorylation site could only be identified in the sample treated with HU (Supplementary Figure S2), indicating that HU treatment induces Ser145 phosphorylation. Based on these results, we hypothesized that PRL1 is phosphorylated by WEE1 at Ser145. To test this hypothesis, we substituted Ser145 to non-phosphorylatable Ala (S145A) and performed in vitro phosphorylation assays. In contrast to MBP-PRL1, the phosphorylation level of MBP-prl1Ser145A was not enhanced by adding GST-WEE1 into the reaction, indicating that WEE1 phosphorylates PRL1 at Ser145 (Figure 4D).
WEE1 promotes PRL1 ubiquitination
Our genetic data suggest that WEE1 negatively regulates PRL1 and our biochemical data suggest that WEE1 phosphorylates PRL1. Next, we aimed to address how WEE1 inhibits the function of PRL1 through phosphorylation. It was reported that phosphorylation promotes protein ubiquitination and subsequent degradation in many cases (43). Therefore, we hypothesized that WEE1-mediated PRL1 phosphorylation triggers PRL1 ubiquitination and degradation. To test this hypothesis, we first examined whether WEE1 affects PRL1 ubiquitination through semi in vitro ubiquitination assays. The recombinant MBP-PRL1 proteins bound to amylose resins were incubated with Col-0 or wee1 extracts, followed by western blotting analysis. It was found that PRL1 could be strongly ubiquitinated by Col-0 extracts, while it was only slightly ubiquitinated by wee1 extracts (Figure 5A). In addition, HU treatment could enhance MBP-PRL1 ubiquitination in Col-0, but not in wee1 extracts (Figure 5A).
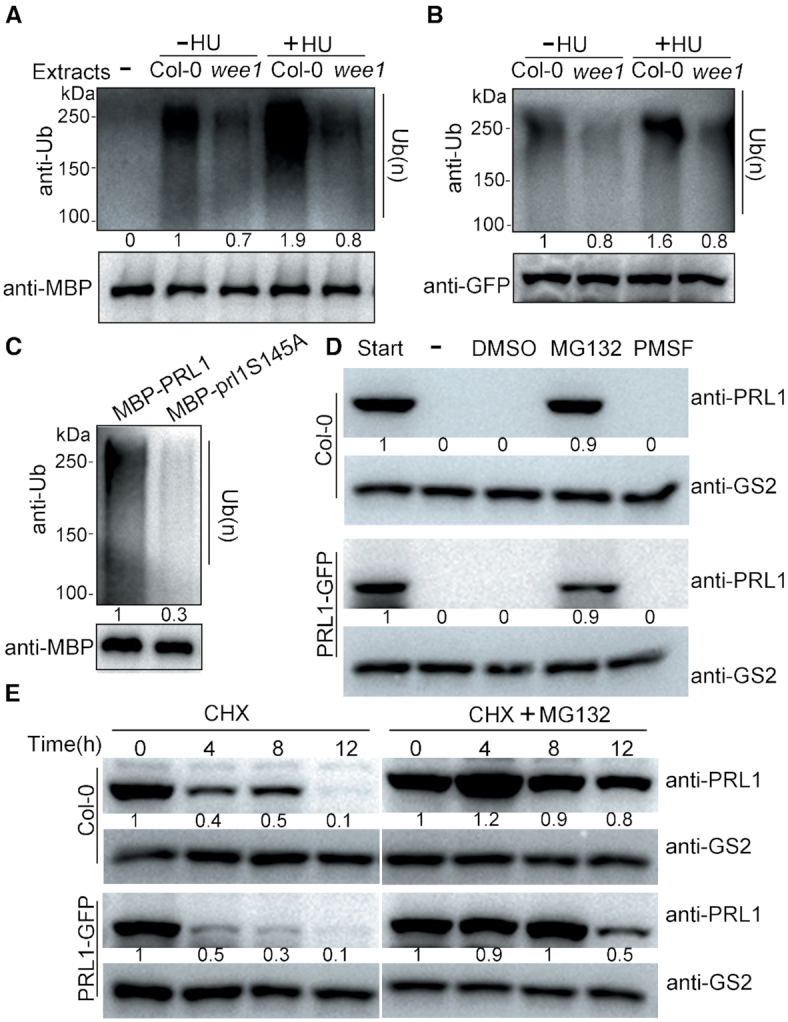
WEE1 promotes PRL1 ubiquitination. (A) In vitro ubiquitination assays. The recombinant MBP-PRL1 protein coupled to amylose resin was incubated with the extracts of Col-0 and wee1 treated with (+) or without (−) 1 mM HU. The resin was subjected to western blotting using anti-ubiquitin (anti-Ub) and anti-MBP antibodies. Ub(n) indicates the ubiquitinated proteins. (B) In vivo ubiquitination assay. The PRL1-GFP proteins were immunoprecipitated using GFP-Trap from the transgenic PRL1-GFP plants in Col-0 and wee1 background and then subjected to western blotting using anti-pS/pT and anti-GFP antibodies. (C) In vitro ubiquitination assay. prl1S145A indicates the non-phosphorylatable PRL1 with the replacement of Ser145 by Ala. (D) In vitro degradation assays. Total proteins were extracted from Col-0 or the PRL1-GFP transgenic plants. The extracts were untreated (−) or treated with 2% DMSO (vehicle), 100 μM MG132 (a proteasome inhibitor), or 4 mM PMSF (a protease inhibitor). After 4 h of incubation at room temperature, proteins were subjected to western blotting using anti-PRL1 antibody. Anti-GS2 were used to determine the loading amount of extracts. (E) In vivo degradation assays. Col-0 and PRL1-GFP transgenic plants were treated with 100 μM cycloheximide (CHX, a protein biosynthesis inhibitor) or a combination of 100 μM CHX and 100 μM MG132 for different times (0, 4, 8 and 12 h). The samples were analyzed by western blot using anti-PRL1 antibody. All experiments were repeated three times with similar results.
To examine whether WEE1 regulates PRL1 ubiquitination in vivo, we tested the ubiquitination levels of PRL1 in the transgenic PRL1-GFP plants. The PRL1-GFP proteins were immunoprecipitated by GFP-Trap and subjected to western blotting. In line with the in vitro data, we found that the ubiquitination level of PRL-GFP was much stronger in Col-0 than in wee1, and HU treatment obviously enhanced PRL1-GFP ubiquitination in Col-0, but not in wee1 (Figure 5B). These results revealed that WEE1 is required for PRL1 ubiquitination, indicating that WEE1-mediated PRL1 phosphorylation is required for its ubiquitination. Since WEE1 phosphorylates PRL1 at Ser145, we tested this possibility by using MBP-prl1S145A for ubiquitination assays. Compared to MBP-PRL1, the ubiquitination level of the MBP-prl1S145A protein indeed reduced dramatically (Figure 5C). Together, these results suggested that WEE1-mediated PRL1 phosphorylation facilitates PRL1 ubiquitination.
PRL1 is degraded through 26S proteasome
PRL1 can be ubiquitylated, indicating that PRL1 may be degraded through 26S proteasome. To confirm this, we performed a cell-free degradation assay using Col-0 or PRL1-GFP transgenic plants. Both the endogenous PRL1 and PRL1-GFP were completely degraded within 4 h (Figure 5D). To distinguish whether the degradation is through protease or proteasome activity, we added the proteasome inhibitor MG132, the protease inhibitor PMSF, or the DMSO solvent in the degradation buffer. Only MG132 could prevent PRL1 and PRL1-GFP degradation (Figure 5D), indicating that PRL1 degradation specifically requires proteasome activity.
Next, we performed in vivo degradation assays by treating Col-0 and the PRL1-GFP transgenic plants with protein biosynthesis inhibitor cycloheximide (CHX) followed by western blotting. As shown in Figure 5E, both the endogenous PRL1 and PRL1-GFP protein levels decreased gradually after CHX treatment and were almost undetectable after 12 h. However, if the plants were co-treated with CHX and MG132, the degradation of PRL1 and PRL1-GFP was largely inhibited. Therefore, both in vitro and in vivo data suggested that PRL1 is subject to 26S proteasome-mediated degradation.
WEE1 is required for PRL1 degradation
To determine whether WEE1 is required for PRL1 degradation, we compared the PRL1 degradation rate in Col-0 and wee1. The recombinant MBP-PRL1 proteins were incubated with the extracts of Col-0 or wee1 to allow for degradation. As shown in Figure 6A, the MBP-PRL1 protein level decreased faster in Col-0 than in wee1, indicating that WEE1 is required for PRL1 degradation in vitro. To examine the in vivo situation, we treated Col-0 and the wee1 mutant with CHX to block protein biosynthesis and then tested the endogenous PRL1 level. Consistently, the endogenous PRL1 in the wee1 mutant was more stable than in Col-0 (Figure 6B).
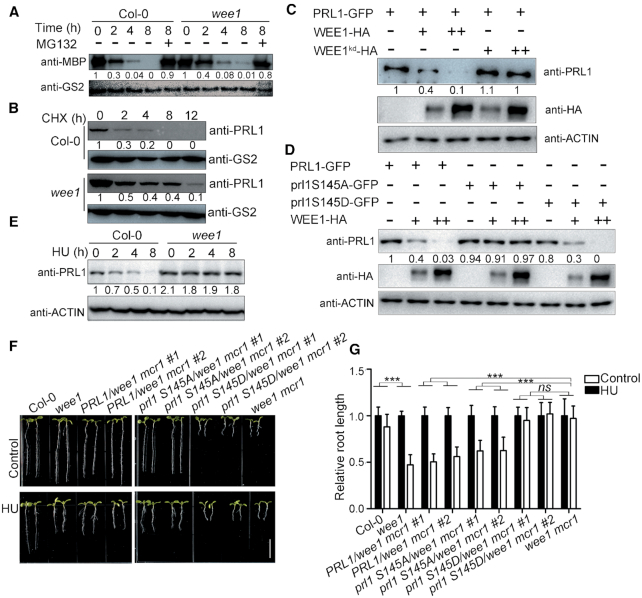
WEE1 promotes PRL1 degradation. (A) In vitro degradation assays. The recombinant MBP-PRL1 proteins were incubated with extracts of Col-0 or wee1. MG132, a proteasome inhibitor. PRL1 was detected using anti-MBP antibody. Anti-GS2 was used to determine the loading amount of extracts. (B) In vivo degradation assays. Col-0 and wee1 were treated with 100 μM CHX (a protein biosynthesis inhibitor) for different times (0, 2, 4, 8 and 12 h). PRL1 was detected with anti-PRL1 antibody. (C and D) WEE1-HA, wee1kd-HA, PRL1-GFP, prl1S145A-GFP, or prl1S145D-GFP was expressed in Arabidopsis protoplasts. WEE1-HA, but not wee1kd-HA, reduced PRL1-GFP protein level when they were co-expressed with PRL1-GFP (C). WEE1-HA reduced the protein levels of PRL1-GFP and prl1S145D-GFP, but prl1S145A-GFP (D). Anti-ACTIN was used as a loading control. (E) Col-0 and wee1 were treated with HU for different times (0, 2, 4, and 8 h). The total proteins were subjected to western blotting. Anti-ACTIN was used as a loading control. (F, G) PRL1 and prl1S145A, but not prl1S145D rescued the wee1 mcr1 mutant. PRL1, prl1S145A, and prl1S145D driven by PRL1 native promoter were transformed into the wee1 mcr1 mutant. The plants were grown vertically on 1/2 MS medium with or without 0.3 mM HU (F and G) for 8 days. The pictures (F) and the relative root length (G) of the indicated plants were shown. Data represent means ± SD (n = 10). Bars = 1 cm. The statistical significance was determined by two-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significance. All experiments were repeated three times with similar results.
To further study the relationship between WEE1 and PRL1 degradation, we tested the PRL1-GFP level in the presence or absence of WEE1-HA in Arabidopsis protoplasts. As shown in Figure 6C, the amount of PRL1-GFP was negatively correlated with the amount of WEE1-HA. Importantly, co-expression of kinase-dead WEE1 (wee1kd-HA) did not affect the level of PRL1-GFP, indicating that WEE1-mediated PRL1 phosphorylation is required for its degradation. In line with this hypothesis, co-expression of WEE1-HA reduced the levels of PRL1-GFP and prl1S145D–GFP, but not prl1S145A-GFP (Figure 6D). These results suggested that WEE1-mediated PRL1 phosphorylation is required for PRL1 degradation.
As shown above, HU treatment enhanced the phosphorylation and ubiquitination of PRL1. Therefore, we proposed that HU treatment enhances PRL1 degradation. Indeed, the endogenous PRL1 protein level decreased after HU treatment (Figure 6E). This is likely due to enhanced degradation because the PRL1 transcript level did not change significantly (Supplementary Figure S3). In the wee1 mutant, the PRL1 level remained stable, indicating that the HU-induced PRL1 degradation is also dependent on WEE1.
To investigate the physiological significance of PRL1 phosphorylation and degradation, we carried our complementation experiment. We transformed PRL1, prl1S145A, and prl1S145D driven by its native promoter into wee1 mcr1 mutants, respectively (Supplementary Figure S4). As shown in Figure 6F and G, while PRL1 and prl1S145A could complement the wee1 mcr1 mutant in terms of both root length and HU response, prl1S145D could not. These results suggested that prl1S145D is constitutively degraded in plants.
PRL1 regulates RNA splicing of cell cycle genes
Our genetic and biochemical data support that ATR-WEE1 negatively regulates PRL1 or the MAC complex. The next question was how loss of function of PRL1 suppresses atr and wee1. In animals and yeasts, ATR and WEE1 function to inhibit cyclin-dependent kinases (CDKs), the major driver of the cell cycle progression. The hypersensitivity of atr and wee1 to HU is partially due to hyperactivation of CDKs. Since the main function of the MAC complex is to regulate RNA splicing (16,27,44,45), we hypothesized that loss of function of PRL1 may result in reduced activities of CDKs through alternative splicing.
In Arabidopsis, PRL1 has one homolog, PRL2, whose expression level is much lower than PRL1 (46). While loss of function of PRL1 causes severe growth defects, the prl2 mutant does not exhibit altered phenotypes, indicating that PRL1 plays a more important role than PRL2 (46). Recently, a total of 1466 genes were found to have intron retention defects in the prl1 prl2 mutant (22). Among these genes, 43 genes are related to cell cycle through gene ontology analysis (Supplemental Data Set 1). Strikingly, CDKA;1 (AT3G48750), CYCD1;1 (AT1G70210) and CYCD3;1 (AT4G34160) are known to play crucial roles in cell cycle progression (47–51). According to the RNA-seq data, the last intron of CDKA;1, the first intron of CYCD1;1, and the first and the third introns of CYCD3;1, showed significantly higher retention ratio in prl1 prl2 mutant than in Col-0 (Figure 7A, B and Supplementary Figure S5). Therefore, we examined the alternative splicing patterns of these three genes using qRT-PCR in Col-0, wee1, mcr1 and wee1 mcr1 before and after HU treatment. As shown in Figure 7C, the first intron retention of CYCD1;1 was induced by HU treatment in Col-0. However, this inducibility was abolished in wee1, indicating that HU-induced intron retention is dependent on WEE1. In the mcr1 and wee1 mcr1 mutants, the intron retention was constitutive. The third intron retention pattern of CYCD3;1 was similar to CYCD1;1. However, we could not reproducibly detect the intron retention pattern of CDKA;1.
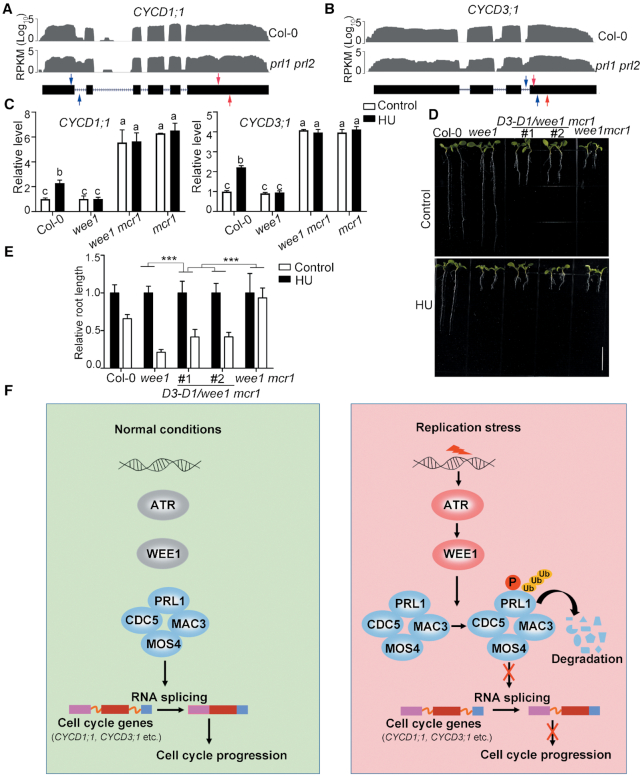
PRL1 modulates alternative splicing of cell cycle genes during replication stress. (A and B) The intron retention defects of CYCD1;1 and CYCD3;1 in prl1 prl2 determined by RNA-seq analysis in previous reports (22). The representative gene model is shown below (black boxes indicate exons and dashed lines indicate introns). The read number is shown in the Y axis. RPKM, reads per kilobase per million mapped reads. (C) qRT-PCR analysis of intron retention events of CYCD1;1 and CYCD3;1 in Col-0 and the indicated mutants. The primer positions are indicated by red and blue arrows in (A and B). The regions between red arrows were used as normalizer. Data represent mean ± SEM (n = 3). The statistical significance was determined using One-way ANOVA analysis with Tukey's multiple comparisons, and different letters (a, b or c) indicated statistical differences (P < 0.01). (D and E) Co-expression of the CYCD1;1 and CYCD3;1 coding sequences could partially restore the growth defects and HU response in the wee1 mcr1 mutant. D3-D1/wee1 mcr1 indicates the transgenic plants co-expressing CYCD1;1 and CYCD3;1 in wee1 mcr1. The plants were grown vertically on 1/2 MS medium with or without 0.75 mM HU for 8 days. The picture (D) and the relative root length (E) of the plants are shown. Data represent mean ± SD (n = 10). Bar = 1 cm. The statistical significances were determined using Two-way ANOVA analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ns, no significance. All experiments were repeated three times with the similar results. (F) A working model to illustrate how the ATR-WEE1 module and the MAC complex regulate replication stress responses. Under normal conditions, the PRL1-containing MAC complex mediates splicing of cell cycle genes including CYCD1;1 and CYCD3;1 to promote cell cycle progression. Upon replication stress, ATR is activated to induce WEE1, which further phosphorylates PRL1 and promotes its ubiquitination and degradation. Without the functional MAC complex, the splicing of cell cycle genes is affected, resulting in cell cycle arrest to allow cells to have enough time to resolve the replication stress.
The intron retention of CYCD1;1 and CYCD3;1 results in premature termination of translation (Supplementary Figure S6). While the full-length of CYCD1;1 encodes a protein with 339 amino acid residues, the one with the first intron encodes a protein with only 120 amino acid residues. The intron-containing CYCD3;1 encodes a protein lacking the last 107 amino acids including Ser 343, which was reported to be a key residue for its function (49). The aberrant CYCD1;1 and CYCD3;1 protein will affect their functions and thus may indirectly reduce the activity of CDKA;1. To test this hypothesis, the coding sequences of CYCD3;1 and CYCD1;1 were co-expressed in wee1 mcr1 using the IntF2A-based polyprotein transgene system (39) (Supplementary Figure S7A–F). As shown in Supplementary Figure S7D, both CYCD3;1 and CYCD1;1 could be detected by western blot. Interestingly, co-expression CYCD3;1 and CYCD1;1 not only partially restored the root length of wee1 mcr1 mutant under normal conditions but also partially restored the HU response (Figure 7D and E). It is to be noted that co-expression of CYCD3;1 and CYCD1;1 in Col-0 neither affected root development nor changed the HU response (Supplementary Figure S7E and F). These results indicate that the HU insensitivity of wee1 mcr1 is partially attributed to the intron retention of CYCD3;1 and CYCD1;1. Taken together, these data suggest that PRL1 can regulate cell cycle by regulating RNA splicing of cell cycle genes.
DISCUSSION
Based on our data, we proposed a working model (Figure 7F). Under normal conditions, the PRL1-containing MAC complex regulates splicing of cell cycle genes including CYCD1;1 and CYCD3;1 to promote cell cycle progression. Upon replication stress, ATR is activated to induce WEE1, which further phosphorylates PRL1 and promotes its ubiquitination and degradation. Degradation of PRL1 compromises the function of the MAC complex, leading to intron retention of cell cycle genes. As a consequence, the cell cycle progression was delayed, allowing cells to have enough time to resolve replication stress. According to this model, PRL1 cannot be degraded in the atr or wee1 mutants, and thus the MAC complex functions normally to promote cell cycle progression, which results in replication catastrophe under stress condition. In the atr prl1 or wee1 prl1 mutants, the cell cycle genes cannot be spliced correctly, leading to cell cycle arrest both in normal and stress conditions.
The ATR-WEE1 module plays a central role in replication stress responses in eukaryotes. Compared with studies in yeasts and animals, how the ATR-WEE1 module functions in plants is less understood (52). Previously, genetic screening was performed to study how WEE1 functions in Arabidopsis (53). It was found that loss of function of RNase H2 leads to the substitution of deoxynucleotide with ribonucleotide in DNA, and thus abolishes the need for WEE1 under replication stress. However, whether WEE1 directly regulates RNase H2 remains unknown. In this study, we found that WEE1 phosphorylates PRL1 and promotes PRL1 degradation. Therefore, our study discovered that PRL1 is a key downstream regulator in the ATR-WEE1 pathway, which significantly advances our understanding of replication stress response in plants.
The MAC complex is evolutionarily conserved from yeasts to animals (54,55). In human cells, the PRP19 subunit is required for ATR activation and thus is an upstream positive regulator of ATR (19,20). In this study, our genetic and biochemical data suggest that the PRL1 or the MAC complex functions downstream of ATR to negatively regulate ATR signaling. Therefore, the function of the MAC complex in the ATR pathway is different between animals and plants. Interestingly, we also found that the prl1 or cdc5 mutants are more sensitive to BLM than Col-0 (Figures 1 and 2), suggesting that the MAC complex plays a positive role in DSB repair. It is worthwhile to study how the MAC complex regulates DSB repair. Taken together, our study provides new insights into the functions of the MAC complex in DDR.
CDKs are the central regulators for cell cycle progression. The activities of CDKs are regulated by multiple mechanisms (10,56–60). First, CDKs are regulated by cyclins. Second, the activities of CDKs are inhibited by CDK inhibitors. Third, CDKs are negatively or positively regulated by WEE1 and CDC25 through phosphorylation and dephosphorylation, respectively. While the first and the second mechanisms are conserved in plants, the third mechanism is still controversial because CDKA;1 containing mutations of the conserved Thr14 and Tyr15 residues could fully complement the cdka;1 mutant both under normal condition and replication stress (11). Therefore, it was proposed that WEE1 activates cell cycle arrest independently of the phosphorylation of CDKA;1 (11). However, it is possible that WEE1 inhibits CDKA;1 through phosphorylation of other residues. It is also possible that WEE1 targets other CDKs or indirectly regulates CDKs by phosphorylating other substrates. In this study, we demonstrated that WEE1 phosphorylates and inhibits PRL1 to regulate alternative splicing of CYCD1;1 and CYCD3;1, which may represent a new cell cycle control mechanism. Since both ATR-WEE1 and the MAC complex are highly conserved in plants, it likely that this mechanism is functional in other plant species. However, since co-expression of the coding sequences of CYCD3;1 and CYCD1;1 in the wee1 prl1 can only partially restore the root length and HU response, it is possible the alternative splicing of other genes also contributes to the prl1 phenotypes.
Although it was reported that WEE1 activates G2/M checkpoint both in animals and plants (61–63), a recent study found that WEE1 also regulates the G1/S regulatory machinery in animals through a haploid genetic screen (64). In further support of this notion, CYCD1;1 and CYCD3;1 were reported to regulate both the G1/S and the G2/M transition. The mRNA of CYCD3;1 peaks at both G1/S and G2/M transitions (65). In addition, overexpression of Antirrhinum majus CYCD1;1 could accelerate cells entry into S phase and M phase in tobacco BY-2 cells (66). Therefore, in the context of replication stress responses, the WEE1-PRL1-CYCDs module is likely to control both the G2/M transition and the G1/S transition.
ACKNOWLEDGEMENTS
We are grateful to Dr Jia Li for providing the pBASTA-AT2 vector, Dr Xia Li for providing mcr1 seeds, Dr Diqiu Yu for providing cdc5 seeds, Dr Guodong Ren for providing wee1 seeds and Dr Bin Yu for pPRL1: PRL1-GFP vector.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31571253, 31771355, 31800216, 31970311]; Fundamental Research Funds for the Central Universities [2662019PY029]; Thousand Talents Plan of China-Young Professionals Grant; and Huazhong Agricultural University Scientific & Technological Self-innovation Foundation [2014RC004]. Funding for open access charge: National Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
1.
2.
3.
4.
5.
6.
7.
9.
10.
11.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
62.
63.
64.
65.
66.