 An Experimental Test of Adaptive Introgression in Locally Adapted Populations of Splash Pool Copepods
An Experimental Test of Adaptive Introgression in Locally Adapted Populations of Splash Pool Copepods
Molecular Biology and Evolution



- Altmetric
As species struggle to keep pace with the rapidly warming climate, adaptive introgression of beneficial alleles from closely related species or populations provides a possible avenue for rapid adaptation. We investigate the potential for adaptive introgression in the copepod, Tigriopus californicus, by hybridizing two populations with divergent heat tolerance limits. We subjected hybrids to strong heat selection for 15 generations followed by whole-genome resequencing. Utilizing a hybridize evolve and resequence (HER) technique, we can identify loci responding to heat selection via a change in allele frequency. We successfully increased the heat tolerance (measured as LT50) in selected lines, which was coupled with higher frequencies of alleles from the southern (heat tolerant) population. These repeatable changes in allele frequencies occurred on all 12 chromosomes across all independent selected lines, providing evidence that heat tolerance is polygenic. These loci contained genes with lower protein-coding sequence divergence than the genome-wide average, indicating that these loci are highly conserved between the two populations. In addition, these loci were enriched in genes that changed expression patterns between selected and control lines in response to a nonlethal heat shock. Therefore, we hypothesize that the mechanism of heat tolerance divergence is explained by differential gene expression of highly conserved genes. The HER approach offers a unique solution to identifying genetic variants contributing to polygenic traits, especially variants that might be missed through other population genomic approaches.
Introduction
As the climate warms, populations of animals and plants that have adapted to historical climates will experience strong selection to respond to new temperature regimes (Aitken et al. 2008; Atkins and Travis 2010; Hoffmann and Sgrò 2011; Shaw and Etterson 2012; Alberto et al. 2013; Kenkel et al. 2013; Rodríguez-Trelles et al. 2013). Current rates of dispersal and plasticity may not be sufficient to keep pace with predicted rates of temperature change (Sunday et al. 2015) and persistence will therefore depend on the species’ capacity for evolutionary adaptation. Rapid adaptation to changing climates has been observed in several species (Bradshaw and Holzapfel 2001; Franks et al. 2007), but current rates of change may outpace adaptive evolution in situ leading to population declines (Møller et al. 2008; Smale et al. 2019). However, in populations that are locally adapted to current gradients in temperature, evolutionary rescue may be facilitated by gene flow, with warming temperatures favoring alleles from equatorward or lower elevation populations (Davis and Shaw 2001). As a result, “rear edge” populations are thought to be an important reservoir of genetic variation necessary for adaptation to warming (Hampe and Petit 2005).
Yet even when genetic variation for adaptive responses to warming exists within a species, adaptive introgression may be hampered by several important genomic factors. First, warm-adapted alleles may arrive to the new population from a genetic background that is not otherwise well adapted to the new location (Schiffers et al. 2012). For example, if local adaptation has occurred along multiple environmental axes, equatorward genotypes may contain warm-adapted alleles that are favored by climate change, but would also contain alleles that would be maladaptive caused by adaptation to contrasting environmental conditions, such as salinity or pH differences among sites. Maladaptation of foreign genotypes may also stem from intralocus selection, where negative epistatic interactions cause migrant alleles to be disfavored in the predominant genetic background of the new population. Second, the power of poleward gene flow to promote evolutionary rescue will be weakened when the trait under selection is highly polygenic. Introgression of a single beneficial allele can happen quickly with strong selection (Miao et al. 2016; Oziolor et al. 2019). However, the selection strength is distributed across each locus of a polygenic trait, potentially weakening the efficiency of introgression (Sachdeva and Barton 2018b). Thus, the probability of evolutionary rescue through gene flow will depend on 1) the architecture of the trait under selection, 2) the degree of maladaptation of the immigrant genetic background, and 3) the rate of recombination, which will determine the capacity of the warm-adapted alleles to escape before being removed by selection against linked maladaptive alleles. Although these factors have been discussed in the theoretical literature (Sachdeva and Barton 2018a, 2018b), few studies have empirically examined the dynamics of introgression for complex traits that are likely to be under selection in changing environments (Zhang et al. 2020). Studies of introgression in natural populations suggest that selection against foreign alleles is often pervasive throughout the genome (Kovach et al. 2016; Matute et al. 2020). However, in cases where there is positive selection on introgressed alleles, this pattern of selection may be repeatable among replicate hybrid populations (Bay et al. 2019).
Here, we examine the process of adaptive introgression using the intertidal copepod, Tigriopus californicus, a small crustacean found in splash pools along the Pacific coast of North America. These shallow pools experience large fluctuations in temperature, providing a unique opportunity to intensively study T. californicus for its physiological adaptations to temperature. T. californicus populations are also highly differentiated with respect to heat tolerance across a geographic range that spans >27° of latitude, suggesting that, at the species level, there is ample genetic variation to adapt to warming temperatures. However, T. californicus populations are also highly subdivided, with individual populations containing ∼1% of the quantitative genetic variation for heat tolerance found at the species level (Kelly et al. 2012). Laboratory selection experiments indicate that individual populations can evolve only ∼0.5 °C additional heat tolerance before exhausting the standing genetic variation available within populations. A species like T. californicus, with local adaptation to temperature and limited variation within populations, would seem to be an excellent candidate for evolutionary rescue through gene flow, or managed translocations, from warm-adapted populations. However, decades of research have also demonstrated that this species exhibits strong outbreeding depression, driven in part by mitonuclear incompatibilities in interpopulation crosses (Ellison and Burton 2010; Willett 2012). For example, hatching success, offspring survival, and successful metamorphosis declines in hybrids of increasing geographic distance between populations along the West Coast of the United States (Edmands 1999).
We evaluated the likelihood of evolutionary rescue via gene flow from the more heat-tolerant equatorward population in the face of mitonuclear incompatibilities. To do this, we hybridized two populations with divergent heat tolerance limits and mitochondrial backgrounds and subjected subsequent generations to strong heat selection. Because we crossed northern females with southern males, all hybrids inherited their mitochondria from the cool-adapted Northern population. As a result, all hybrids were expected to experience selection for higher frequencies of Northern nuclear alleles to alleviate mitonuclear incompatibilities. However, in hybrids under selection for increased heat tolerance, warm-adapted alleles from the Southern population should also experience positive selection. Thus, depending on the number of loci involved, loci across the hybrid genome might experience one or both of these contrasting selection pressures through linkage to loci contributing either to heat tolerance or to mitonuclear incompatibilities.
We used whole-genome resequencing to identify loci responding to heat selection via a change in allele frequency. Evolve and resequence methods combine experimental evolution (Kawecki et al. 2012) with sequencing to identify the targets of selection. This approach has been used to map the genetic basis of a variety of traits in Drosophila, including body size (Turner et al. 2011) and courtship song (Turner and Miller 2012), but we are aware of no study to date that has applied this approach to a marine metazoan. In addition, hybridization of divergent populations, or species, combined with Evolve and Resequencing has been performed in many systems, such as Drosophila (Matute et al. 2020) and yeast (Burke et al. 2014; Zhang et al. 2020). To characterize this field of study, we propose the acronym hybridize evolve and resequence, or HER (modified from “Evolve and Resequence”; Turner et al. 2011).
Results
Increased Heat Tolerance in Selected Lines
We created hybrid lines by crossing 20 females from Bodega and 20 males from San Diego (fig. 1). These two populations have different thermal tolerances, with median lethal (LT50) temperatures at 34.8 and 36.5 °C for Bodega and San Diego, respectively (fig. 1). The F1 generation of hybrid crosses had an LT50 value in between that of the two parental populations, with the direction of the cross having no influence on the LT50 value (i.e., ♀ Bodega and ♂ San Diego crosses vs. ♀ San Diego and ♂ Bodega crosses; fig. 1). Starting with the F2 generation (using ♀ Bodega and ♂ San Diego crosses), we established five replicate “selection” lines and “control” lines. Each generation, selected lines were exposed to the temperature that produced 50–90% mortality. After 15 generations of heat selection in the selected lines, hybrids had a significantly higher LT50 temperature than control lines (F1,18 = 86.6, P < 0.001; fig. 1) with selected lines exhibiting a ∼1.2 °C higher tolerance. The difference in LT50 temperatures between our selected and control lines demonstrate that we successfully selected for increased heat tolerance in hybrid copepods. Additionally, hybrids from the selected lines either exceeded or were within ∼0.5 °C of the San Diego population LT50 temperature. Copepods from the control lines had lower LT50 than selected lines and the F1 generation, suggesting that they had lost heat tolerance alleles from the San Diego population in favor of Bodega alleles to alleviate mitonuclear incompatibilities.

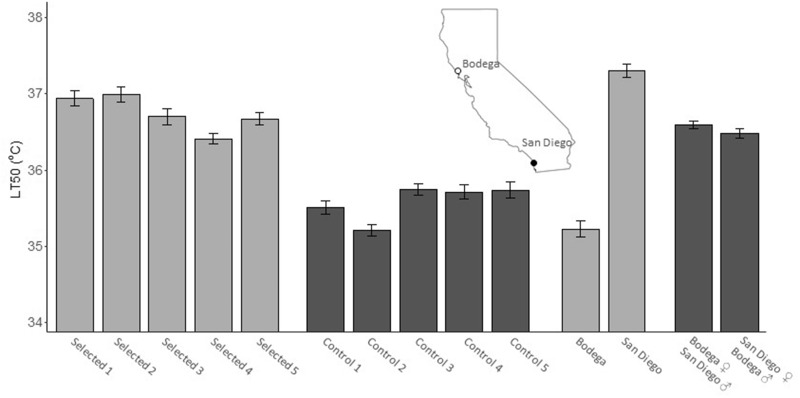
Median lethal temperatures (LT50) for hybrid lines selected for heat tolerance (Selected 1–5) and controls lines 1–5. Parental copepods from the pure Bodega and San Diego population are displayed as well as the F1 hybrid generation crossed as either Bodega–San Diego or San Diego–Bodega (maternal–paternal, respectively).
Whole-Genome Resequencing
We generated 12 whole-genome libraries (five selected lines and five control lines plus one sequencing library for each of the pure parental populations) for pool-sequencing and aligned reads to the T. californicus genome version 2.1 (created from the San Diego population; Barreto et al. 2018). Mean mapping success of all libraries was 64.2% (supplementary table S1, Supplementary Material online). The San Diego population had the highest mean mapping rate (74.8%) as expected since the genome was assembled from the same population, whereas the Bodega population had a mean mapping rate of 61.0%. The selected and control lines had a mean mapping rate of 66.0% and 61.0%, respectively. We also created a Bodega pseudoreference genome using SNPs identified in a Bodega VCF file. Mapping rates for all libraries to the Bodega reference were similar or slightly higher than mapping rates to the San Diego assembly (supplementary table S1, Supplementary Material online). The following analyses are based on mapping to the San Diego genome, but we also compare our results between the two population-specific reference genomes to show that mapping rates do not result in biased estimates of allele frequencies (see Results and Supplementary Material online).
Consistent Allele Frequency Changes among Selected Lines
A PCoA of genome-wide patterns in allele frequencies shows that samples cluster by selection or control treatment (Adonis P < 0.01), suggesting consistent shifts in response to treatment conditions (fig. 2). Samples also cluster closer to the pure Bodega population confirming our expectation that across most of the genome, mitonuclear incompatibilities should favor nuclear Bodega alleles compatible with the Bodega mitochondrial background.
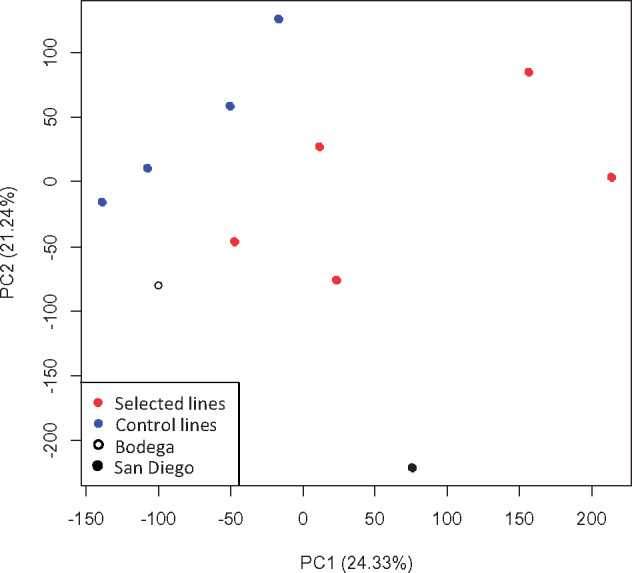
Principle coordinate analysis of allele frequencies for selected lines (red), control lines (blue), the pure Bodega population (white), and the pure San Diego population (black).
To identify regions of the genome under selection for heat tolerance, we estimated allele frequency differences across selected lines relative to controls. In the F1 generation, all lines begin with a 50/50 ratio of San Diego and Bodega alleles. In the absence of selection, these allele frequencies are expected to depart from their initial frequencies due to drift only. However, in T. californicus, mitonuclear incompatibilities will select for nuclear Bodega alleles that are compatible with the Bodega mitochondrial alleles, causing allele frequencies to depart from expectations under drift. Both selected and control lines will be experiencing mitonuclear selection, but only selected lines will be experiencing heat selection. Therefore, reads from control lines were pooled so we could individually compare each selected line to all control lines to identify changes in allele frequencies only due to heat tolerance selection relative to allele frequencies generated by drift and intralocus selection in controls. We used a Cochran–Mantel–Haenszel (CMH) test to estimate the likelihood of consistent changes in allele frequencies across all replicate selected lines in comparison to the pooled control line across a 10,000-bp window. After our Bonferroni correction (P = 0.05/18,796 windows), there were 8,083 windows that had a significant change in allele frequency across all selected lines compared with the pooled control line (windows above blue horizontal line in fig. 3A). These windows spanned all 12 chromosomes and are strong candidates for loci that are under selection for heat tolerance. A CMH test was also performed on reads mapped to the Bodega reference genome (supplementary fig. S1A, Supplementary Material online). The two CMH tests shared similar distributions and there were a total of 1,198 shared windows that passed the significance threshold for reads mapped to both reference genomes.
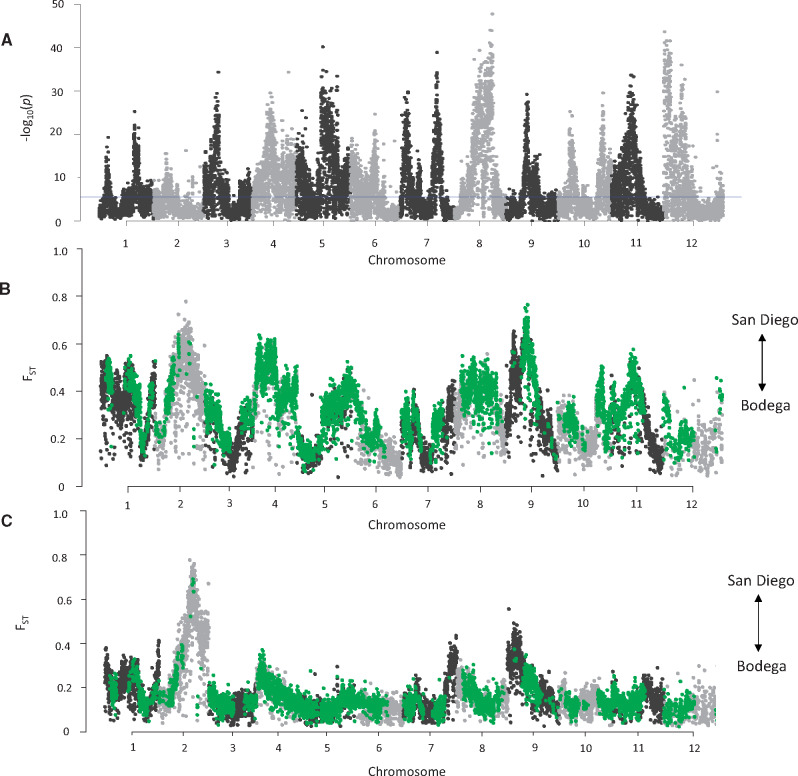
(A) Manhattan plot showing 10,000-bp windows that had consistent changes in allele frequencies across all selected lines compared with pooled control lines. P values are displayed as −log from CMH test. The blue horizontal line represents our significance threshold after Bonferroni correction. (B) FST average of 10,000-bp window of all selected lines versus the pure Bodega population. (C) FST average of 10,000-bp window of all control lines versus the pure Bodega population. Windows colored green on FST plots are loci that were above our Bonferroni correction from (A).
Southern Alleles under Selection for Heat Tolerance
To determine if regions identified as under selection for increased heat tolerance contained alleles that originated from the San Diego population, we estimated FST between the selected lines and the two pure populations. Regions with high FST for selected versus Bodega, but not control versus Bodega, are expected to be loci under selection for heat tolerance, since heat tolerance selection should mostly favor alleles from the heat-tolerant (San Diego) population. Since all copepods have a Bodega maternal line, mitonuclear incompatibilities should favor nuclear Bodega alleles compatible with the Bodega mitochondrial background. Regions with low FST relative to Bodega in both the control and the selected lines are expected to be loci with high mitonuclear incompatibilities, or loci where the Bodega allele had higher fitness under laboratory conditions. We first compared the genome-wide mean FST for selected and control lines to the Bodega pure population so that loci of high FST corresponded to a higher frequency of alleles from the heat-tolerant San Diego population. The overall genome-wide mean FST for selected lines versus the pure Bodega population was higher (0.31; fig. 3B) than control lines versus Bodega (0.17; fig. 3C). This matches our expectations that the selected lines will have a higher frequency of alleles from the heat-tolerant San Diego population and control lines will have a higher frequency of alleles from the Bodega population that would be compatible with the Bodega mitochondrial genes (further evidence for mitonuclear incompatibilities that exist between these two populations). Windows identified to be under heat tolerance selection in the selected lines (from fig. 3A) also had higher FST values compared with the pure Bodega population (0.35) than control lines compared with pure Bodega (0.15) (fig. 3B and C represented as green dots). Thus, windows that had high allele frequency change between selected and control lines (fig. 3A) also had high FST values when comparing the selected lines to the pure Bodega population (fig. 3B). We found the reverse to also be true when comparing the selected and control lines to the pure San Diego population. The selected lines had a lower genome-wide mean FST value (0.37; supplementary fig. S2A, Supplementary Material online) compared with the pure San Diego population than the genome-wide mean FST for control lines (0.55; supplementary fig. S2B, Supplementary Material online). Similarly, windows under heat tolerance selection had a lower FST value in the selected lines (0.33; supplementary fig. S2A, Supplementary Material online) than the control lines (0.62; supplementary fig. S2B, Supplementary Material online), meaning windows under selection were more similar to the San Diego population in the selected lines than the control lines. These results were confirmed in a comparison of allele frequencies between selected and control lines: selected lines had a higher mean frequency of San Diego alleles for SNPs under heat tolerance selection (supplementary fig. S3, Supplementary Material online).
The Manhattan plots of both the selected and the control lines also allow us to make an interesting investigation of universally beneficial alleles. For example, there were loci with high FST values relative to the Bodega population on chromosomes 1, 2, 7, and 9 in both the selected and the control lines. These regions of universal high FST (i.e., higher frequency of San Diego alleles) may provide a fitness advantage under laboratory conditions.
Selection Coefficients and Recombination Rates Are Low
We created a simple model to explore what combinations of selection coefficients and recombination rates might yield the F16 allele frequencies observed in our experiment. Model calculations and output can be seen in supplemental file 1. Our model yields several observations consistent with what we know about the biology of Tigriopus: 1) recombination rates must be low. With higher rates of recombination, greater selection against incompatibility alleles is needed than under lower rates of recombination to yield the final allele frequencies. If, as we suspect, incompatibility effects are recessive, then recombination between heat tolerance and incompatibility alleles cannot be greater than 4% per generation (supplementary fig. S4, Supplementary Material online), because an r of 0.04 requires that selection on incompatibility alleles to be equal to −1, meaning that recessive alleles are lethal in their homozygous form. 2) Selection coefficients for heat tolerance alleles are smaller in magnitude than selection coefficients for incompatibility alleles (supplementary fig. S4, Supplementary Material online), but greater values of selection (for heat tolerance alleles) are required to generate the observed allele frequencies under lower recombination rates (s = 0.22 for r = 0.004) than under higher recombination rates (s = 0.12 for r = 0.04). This is consistent with what we know about the biology of heat tolerance as a polygenic trait: Given that there are many loci under selection in our heat tolerance lines, we expect the selection coefficient for any one locus to be relatively modest. Finally, we expect the realized selection coefficient for most heat tolerance loci to be negative during the initial phase of introgression if they are linked to incompatibility loci. We can see this when we plot the allele frequencies generated by our model across 15 generations of selection: Across the range of recombination values and selection coefficients that we tested, allele frequencies for heat tolerance alleles initially decline, before increasing again in frequency by generation F16 (supplementary fig. S4, Supplementary Material online).
Genomic Mechanisms of Heat Tolerance
There were 796 genes that overlapped with the windows that were identified as under selection from the CMH test. These genes were enriched in three molecular functions: molecular function regulator, enzyme inhibitor, and DNA binding. However, no categories that might specifically explain heat tolerance were enriched. Our ability to identify specific genes under selection is most likely hampered by high levels linkage disequilibrium (LD) due to low recombination rates. In addition, high levels of LD may be the result of a selective sweep where adjacent windows containing neutral SNPs are swept to high frequencies, thus impeding our ability to identify the actual target of selection. Nevertheless, we can still make some inferences on the mechanism of heat tolerance by investigating sequence divergence and differential gene expression.
To elucidate the mechanism of increased heat tolerance in the selected lines, we tested whether genes under selection for heat tolerance contained genes with high coding sequence divergence between populations. We estimated Ka/Ks values (the ratio of nonsynonymous to synonymous substitutions as an indicator of selective pressures on genes) between populations. The mean Ka/Ks value for the genes under selection was 0.59 which was lower than the genome-wide average Ka/Ks of 0.667 (t16048 = 6.25, P = 4e-10). A permutation test confirmed this significant difference in Ka/Ks values was not due to a small sample size; the mean Ka/Ks value for genes under selection (0.59) fell below the range of the 95% permutation distribution (0.63, 0.67). This result was consistent with reads mapped to the Bodega reference genome: the mean Ka/Ks value for the genes under selection was 0.62 which was lower than the genome-wide average (t1790 = 1.98, P = 0.047). Purifying selection acting on these genes under heat tolerance selection suggests that these genes may be highly conserved in the genome of both populations.
We also tested whether genes under selection were differentially expressed by reanalyzing transcriptomic data from Kelly et al. (2017). In this previous experiment, hybrid individuals were used from the same experimental lines described here and after four generations the number of differentially expressed genes (DEGs) was measured in response to heat shock (a stressful but nonlethal temperature) and ambient conditions for both selected and control lines. We tested for a genotype-by-environment interaction and found that windows under selection for heat tolerance were enriched in genes that have different expression patterns between selected and control lines in response to heat shock (Fisher’s exact test, P = 0.03; table 1). However, this was only true when reads were mapped to the San Diego reference genome (table 1). In addition, windows under heat tolerance selection were enriched in genes that were differentially expressed to heat shock in both the selected and the control lines (Fisher’s exact test, P < 0.001; table 1). This was true for reads mapped to the San Diego and Bodega reference genome. This suggests that genes that are a common response to heat shock (differentially expressed in both selected and control lines) are also under selection for long-term heat tolerance.

| Data Input | Effect | # DEG | No. SD Windows under Selection That Overlap with DEG (FET P value) | No. BR Windows under Selection That Overlap with DEG (FET P value) |
|---|---|---|---|---|
| All | Line × Heat shock | 333 | 102 (P = 0.0304*) | 24 (P = 0.5614) |
| All | Heat shock | 591 | 234 (P < 0.0001*) | 62 (P = 0.0014*) |
| All | Line | 30 | 13 (P = 0.0776) | 6 (P = 0.0201*) |
| Heat shock | Line | 184 | 63 (P = 0.0161*) | 19 (P = 0.0698) |
| No heat shock | Line | 148 | 49 (P = 0.0519) | 12 (P = 0.3984) |
| Selected line | Heat shock | 117 | 37 (P = 0.144) | 11 (P = 0.2544) |
| Control line | Heat shock | 1,981 | 671 (P < 0.001*) | 180 (P = 0.008*) |
Note.—Displayed is the number of DEGs from mapping to the newly available genome (Barreto et al. 2018). Unique DEGs from all the comparisons were used to determine overlap with genes within windows under heat tolerance selection. The P value for the Fisher’s exact test (FET) is displayed in parentheses to test for enrichment for each DEG category. Significant P values < 0.05 are marked with an *.
Discussion
The possibility of evolutionary rescue through gene flow has been discussed in the theoretical literature (Bell 2013; Sachdeva and Barton 2018a, 2018b), but few studies have empirically tested this idea. We demonstrate the capacity of warm-adapted alleles to increase in frequency in hybrid copepods under heat selection in the face of mitonuculear incompatibilities. Using the HER technique, we observed consistent shifts in allele frequencies across replicate lines at multiple loci, suggesting that heat tolerance is a highly polygenic trait. Our results contribute to the growing body of evidence that introgression of complex adaptive traits can occur relatively quickly, even in an otherwise maladaptive genetic background (Kovach et al. 2016; Bay et al. 2019; Oziolor et al. 2019; Matute et al. 2020).
Thermal Tolerance Outweighs Cost of Outbreeding Depression
Previous studies have observed beneficial effects of hybridization between geographically distant populations of T. californicus (Willett 2012). Hybrids typically have a higher thermal tolerance than at least one of the parental populations, suggesting that under stressful environments, the benefits of hybridization outweigh the negative impacts of outbreeding depression. Our results agree with these findings. Not only does the F1 generation of hybrids have a thermal tolerance higher than the cool-adapted Bodega population but the selected lines continue to maintain a high thermal tolerance. This result is also supported by the high frequency of alleles under selection originating from the warm-adapted San Diego population. This suggests that under continued exposure to stressful high temperatures, the negative cost of incompatible gene combinations is outweighed by selection for increased heat tolerance. We see evidence for hybrid breakdown in the control lines that did not experience stressful heat exposure, and as a result, these lines have a higher frequency of nuclear alleles originating from the Bodega population.
Empirical Implications of Adaptive Introgression of Polygenic Traits
Our experiment adds to the limited number of studies that empirically test the adaptive introgression of polygenic traits in the context of climate change. The theoretical literature stresses the difficulty of identifying the actual targets of selection due to hitch-hiking of neutral SNPs in large linkage blocks, especially in the early stages of experimental tests of introgression (Sachdeva and Barton 2018b). However, over longer timeframes, recombination will break up large linkage blocks so selection can act on smaller-scale variation, making it possible to further refine the target of selection. In our experiment, the large size of our linkage blocks (as evident by wide elevated peaks in the CMH and FST plots; fig. 3A and B), suggest that we are still in the early stages of introgression. As a result, we were unable to identify specific SNPs under selection. Further generations of selection should allow recombination to separate the actual target of selection from neutral SNPs swept to high frequencies. Nevertheless, we are able to describe general patterns of the mechanism of heat tolerance that we outline below.
Differential Gene Expression of Highly Conserved Genes May Explain Mechanism of Heat Tolerance
Although we identified a few functions enriched for genes under selection, none were directly related to stress responses. Our inability to detect substantial functional enrichment in genes under selection likely stems from the fact that our data set contains a large number of “false positives”—alleles which increased in frequency due to linkage with loci contributing to heat tolerance but are themselves functionally unrelated to heat tolerance. These loci would have tended to dilute any functional enrichment we might have observed among genes responding to selection. However, we found that genes under selection for heat tolerance have lower genetic divergence (low Ka/Ks values) than the genome-wide average. In addition, these windows under selection were enriched for genes that were differentially expressed in common-response type genes (both selected and control lines changed expression levels in response to short-term heat shock). This suggests that genes that are important for short-term heat shock are also important for long-term adaptation to heat stress. Importantly, these windows under selection were enriched in genes that showed a genotype-by-environment interaction where there were differences in gene expression between selected and control lines in response to heat shock. Thus, the feature of these loci that generates the observed differences in heat tolerance between selected and control lines may be regulatory changes to highly conserved genes. However, we acknowledge that the generation gap between RNA-sequencing (generation four) and whole-genome resequencing (generation 15) prevents us from forming conclusive correlations.
Selection on Polygenic Traits in Natural Populations
Heat tolerance has been demonstrated to be a highly polygenic trait in other species, such as Drosophila (Tobler et al. 2014; Michalak et al. 2019). In T. californicus, we provide evidence for the polygenic nature of heat tolerance by identifying multiple loci that showed consistent shifts in allele frequencies in the selected lines compared to the control lines. We propose that these loci were initially subjected to divergent selection in the parental populations when they were being established in the past. However, polygenic traits are typically modeled as being under long-term stabilizing selection, where the intermediate trait value has the highest fitness (Barton and Keightley 2002). We see potential evidence for long-term stabilizing selection on heat tolerance in T. californicus populations which are locally adapted to their individual thermal regimes (Kelly et al. 2012). It is evident that the species as a whole is capable of adapting to extreme temperatures (populations found near the equator have the highest LT50 values), but fecundity declines with higher heat tolerance (Kelly et al. 2016). Thus, populations have arrived at an optimal thermal lethal limit slightly below the maximum temperature documented at each location (Kelly et al. 2012) to limit the loss in fecundity while also limiting mortality from heat events.
To maintain trait optimums, stabilizing selection on polygenic traits often results in reduced genetic variation at each locus, which may be maintained by purifying selection at the molecular level. This may explain why genes involved in heat tolerance seem to be under purifying selection (low Ka/Ks values) and are highly conserved in our study. When a population is close to its optimum trait value, this often results in reduced genetic variation (Barton 1986; de Vladar and Barton 2014), as deviations from the optimum are selected against.
These results highlight the utility of the HER technique. The two most common approaches to identifying the genetic basis for local adaptation involve scans for loci which show the greatest differentiation among populations, and tests for allele frequencies that show correlations with environmental variables (Hoban et al. 2016). A major challenge to these approaches in the context of polygenetic traits is that selection typically occurs through small changes in allele frequencies at each locus (Yeaman 2015), making it difficult to pinpoint loci under selection. Furthermore, tests of adaptive differentiation are easily confounded by population structure and other covarying sources of selection. The HER technique offers a unique solution since all alleles that differ between populations start at a frequency of 50%. By directly applying a single source of selection, we expect any consistent changes in allele frequencies to be the result of selection on that trait alone. Although our study could be further improved by extending the number of generations to allow more time for recombination to break up the target of selection with linked neutral sites, HER provides a starting point for future studies to expand on. We were able to identify potential regions that are important for heat tolerance, which could be further investigated in conjunction with GWAS or linkage-mapping studies (Hoban et al. 2016).
Conclusions and Future Implications
Our results suggest that evolutionary rescue through introgression from a stress-tolerant population can be a possible avenue to prevent population extirpation from climate change. Individual populations subjected to generations of heat tolerance selection were unable to increase their heat tolerance by more than half a degree (Kelly et al. 2012), but we show that hybrid copepods are able to increase their heat tolerance to levels seen in warm-adapted populations. Heat tolerance alleles can reach high frequencies in hybrid copepods despite the strong mitonuclear incompatibilities developed over years of separation between these two populations. However, we did not test possible fitness tradeoffs in the hybrid individuals in this study even though it has been previously demonstrated that hybrids have reduced fecundity and plasticity (Kelly et al. 2013, 2016). In addition, evolutionary rescue is unlikely to occur naturally in this species due to their limited dispersal. Assisted evolution through breeding populations with stress-tolerance alleles may hold promise for preventing species extinction; however, hybrids will need to compete with the established population. There may be other environmental axes of adaptation that may put the hybrids at a disadvantage, for example, southern populations might have higher salinity tolerances than northern populations (DeBiasse et al. 2018). Thus, further research would be required to determine how long it would take the Bodega population to completely disentangle heat tolerance alleles from potentially maladaptive linked loci. Therefore, the fitness of the hybrids in the wild will depend on the rate of recombination to remove maladapted linked loci as well as the selection pressure on the introduced alleles in the new environment.
Materials and Methods
Field Collection, Hybrid Crosses, and Experimental Selection
Laboratory cultures were established from two populations collected from Bodega Marine Reserve in northern California (38°04′ N, 123°19′ W) and San Diego in southern California (32°49′ N, 117°16′ W) in 2014 (fig. 1). These two populations had the largest difference in heat tolerance that were still compatible. Cultures were maintained at ambient temperature (20 °C) and salinity (35 ppt) under 12-h light/12-h dark conditions and fed ground spirulina fish food ad libitum for two generations (∼ 8 weeks) before initiating crosses.
To create hybrid laboratory populations, 20 females from Bodega and 20 males from San Diego were crossed. Copepods form mate-guarding pairs, in which a male copepod guards the virgin female by attaching to her with specialized antennae. These pairs from each population can be teased apart with a fine probe and then paired with a partner from the opposite population. Newly formed pairs were held in a 24-well tissue culture plate until the female produced her first brood. Most genetic variation for thermal tolerance is partitioned between populations (Kelly et al. 2012), therefore a starting population size of 20 individuals from each population was sufficient to sample the genetic variation within populations. To ensure the direction of our cross did not affect the LT50 temperature for the F1 generation, we also performed a similar cross with males and females from the opposite population, that is, 20 males from Bodega and 20 females from San Diego.
Once the F1 generation (for ♀ Bodega and ♂ San Diego crosses) reached adulthood and began forming mate-guarding pairs, we established five replicate lines with 30 mate-guarding pairs each. Parental and F1 generations were maintained at ambient conditions as described above. Starting with the F2 generation, from each of the five replicate lines, 40 mate-guarding pairs were haphazardly chosen for the “selection” line and 40 pairs for the “control” line. Experimental exposure for the selected lines is described as follows. To select for increased heat tolerance, we exposed the selected lines to the temperature that produced 50–90% mortality for 1 h (36.0 °C in the first generation of selection, increasing by ∼0.1 °C per generation of selection). This initial temperature was determined by calculating the LT50 temperature for the F1 generation hybrid crosses as described in the following section. All copepods from the selected line were exposed using a digital water bath and then we haphazardly sampled 40 of the surviving mate-guarding pairs that then became the parents for the next generation. The control lines were propagated for 21 generations, whereas the selected lines were propagated for a total of 15–16 due to the increased amount of time per generation to achieve adequate numbers of individuals for the selection treatment.
Heat Tolerance Measurements
We determined the LT50 temperatures for each parental population line (Bodega and San Diego), the F1 generation for both hybrid crosses (♀ Bodega and ♂ San Diego; ♂ Bodega and ♀ San Diego), and after 15–21 generations for each selected and control line derived from ♀ Bodega and ♂ San Diego hybrid crosses. For each of these groups, individual mate-guarding pairs were collected and placed into PCR tubes with seawater. For each of the selected and control lines, we exposed sets of nine mate-guarding pairs to a target temperature in a thermocycler for 1 h, allowed 48 h for recovery, and then assessed survival. We did this for a series of 5–10 temperatures at 0.2 °C intervals, spanning from the temperature that produced 90–100% survival to the temperature that produced 90–100% mortality, and then used the mortality at each temperature to estimate LT50 for each line via logistic regression. We pooled results from females and males after determining there were no differences in LT50 scores between sexes. The LT50 for each group was then estimated using a logistic regression and the R package MASS (Venables and Ripley 2002). A two-way ANOVA was performed to test the effects of selection and sex on LT50 temperatures with each line treated as a random factor using the R package nlme (Pinheiro et al. 2019) in R v. 3.4.4.
Whole-Genome Resequencing
One sequencing library was made for each of the five selected and five control lines plus one sequencing library for each of the pure parental populations (12 libraries total). For each library, 100 copepods were pooled and concentrated to remove as much water as possible. DNA was extracted using the Sigma Aldrich kit following manufacturer’s instructions. Yield and quantity were assessed using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). We constructed 12 whole-genome libraries from sonicated DNA pools using the NEBNext Ultra DNA Library Prep Kit for Illumina (Illumina, San Diego, CA), following manufacturer’s instructions. Concentration and fragment size distributions for each library were assessed using a Bioanalyzer (Agilent, Santa Clara, CA). Each library received an individual barcode adapter with five and seven libraries pooled and sequenced on two lanes of an Illumina HiSeq 3000 with a goal coverage of 30×. Paired-end (2 × 150 bp) sequencing was performed at Iowa State University’s DNA Facility (Ames, IA). Low-quality bases and adapters were removed using FastQC v0.11.5 (Andrews 2010). Reads were mapped to the Tigriopus californicus genome version 2.1 generated from the San Diego population (Barreto et al. 2018) using bowtie2 (Langmead and Slazberg 2012). We also mapped reads to a Bodega pseudoreference genome that we created with a VCF file using the Bcftools consensus flag. The Bodega VCF file was created following the GATK pipeline: Cleaned reads from the Bodega population were mapped to the San Diego reference using bwa v0.7.17 (Li and Durbin 2009). Duplicate reads were removed and then remaining reads were indexed using Picard tools. Finally, SNPs and indels were identified using HaplotypeCaller with GATK v4.0.2.1 and compiled into a VCF file.
Identifying Windows under Selection
To visual genome-wide relationships among samples, we performed a principle coordinate analysis (PCoA) and calculated the effect of selection on allele frequencies using the function Adonis in the R package vegan (Oksanen 2015). We followed the pipeline outlined in Popoolation to estimate SNP frequencies in our samples. We estimated nucleotide diversity with the perl script Variance-sliding.pl and a window size of 10,000 bp.
We also followed the pipeline outlined in Popoolation2 (Kofler et al. 2011) for estimation of FST between selected lines and the two pure populations. A synchronized file containing SNP frequencies for each sample was created using the samtools mpileup option and the perl script mpileup2sync.pl through Popoolation2. FST values were calculated using the sliding window approach with the fst-sliding.pl perl script (Kofler et al. 2011). A window size and step size of 10,000 bp was used with a minimum coverage of 10×. FST comparisons were computed for each selected and control line against the two pure populations.
To estimate consistent allele frequency differences across selected lines relative to controls, we first pooled all control line reads into a single fastq file. We then followed the suggested pipeline outlined in Popoolation2 and performed a CMH test to obtain a single P value for consistent changes in allele frequencies across all replicate lines in comparison to the pooled control line. The CMH test calculates P values for SNPs which are assumed to be nonindependent, which greatly inflated the significance. To correct for the large linkage blocks in our experiment, we calculated the geometric mean of P values within a 10,000-bp window. Windows were identified to be under heat tolerance selection after passing the Bonferroni correction threshold for multiple comparisons based on the total number of windows, which was 18,491.
Modeling Selection and Recombination
To explore what combinations of selection coefficients and recombination probabilities might generate our observed results, we created a simple model of selection on two loci plus recombination in hybrids. In our model AS is a southern allele which confers increased heat tolerance, whereas AN is the northern allele at the same locus. We modeled heat tolerance alleles as additive, so that in the heat selection lines, the fitness of ANAN is 1, ANAS is 1 + sA, and ASAS is 1 + 2sA. Locus A is linked to mitonuclear incompatibility locus B. We modeled the effects of mitonuclear incompatibilities as recessive, because the fitness of F1 hybrids in Tigriopus is generally greater than that of either parental genotype, with hybrid fitness declining sharply in the F2 generation, suggesting recessive-acting incompatibility alleles (Willett 2012). Therefore, in our model, the fitness of BNBN is 1, BNBS is 1, and BSBS (which has two alleles incompatible with the northern mitochondrial background) is 1 − sB. Finally, the probability of a recombination event between locus A and B is equal to r.
Each generation, we first calculate the frequency of the four types of gametes (ASBS, ASBN, ANBS ANBN) after recombination, given r and the frequencies of each parental genotype. Next, we calculate the frequency of each of the genotypes in the next generation after random mating, given the frequencies of each type of gamete. Finally, we calculate the new frequencies of each genotype in the next generation after one round of viability selection given the selection coefficients for both loci. We repeated this process 15 times to obtain the final frequencies expected for the AS and BS alleles in the heat selection lines, given sA, sB, and r. In the control lines, we set sA = 0, since there was no selection for heat tolerance, and calculated expected frequencies of AS and BS, given sB and r only. We set F16 allele frequencies for AS to 0.50 in selected lines which was a typical frequency for alleles in the center of windows under selection at F16. We set the frequency of AS to 0.12 in control lines, which was the mean allele frequency of these same alleles in control lines. We then used our model to calculate selection coefficients necessary to generate these observed allele frequencies across a range of recombination probabilities. An excel spreadsheet which performs all of these calculations for different values of sA, sB, and r can be found in Supplemental file 1.
Functional Enrichment and Gene Expression
Windows that passed the Bonferroni correction for allele frequency differences were consequently annotated. For windows that overlap with gene regions, we extracted GO terms from the GFF file and performed a functional enrichment test (Wright et al. 2015) using the Fisher’s exact test option and a false discovery rate correction.
We reanalyzed transcriptomic data from Kelly et al. (2017) due to the availability of a new reference genome (Barreto et al. 2018) and determined the number of differentially expressed genes (DEGs) after four generations of selection for increased heat tolerance. There are four experimental conditions: selected lines exposed to 1 h of heat shock at 34 °C, selected lines maintained at ambient temperature, control lines exposed to 1 h of heat shock at 34 °C, and control lines maintained at ambient temperature. To determine if DEGs were also genes in windows under selection, the transcriptomic and whole-genome data needed to be mapped to the same reference genome. Raw transcriptomic reads were cleaned using TrimGalore v0.4.4, which removed low-quality bases (Q < 20) and adapters with FastQC v0.11.7 (Andrews 2010) and Cutadapt v1.18 (Martin 2011). Cleaned reads were mapped as single end to the reference genome v2.1 (Barreto et al. 2018) using RSEM v1.3.0 (Li and Dewey 2014). DEG analysis was performed using the DESeq2 package (v1.18.1) in R (v3.4.4) using an adjusted P value cut off of 0.05 (Love et al. 2014). We tested the gene expression response to heat shock and ambient temperature for both selected and control lines. Our number of DEGs for different experimental treatments corresponded closely with results from Kelly et al. (2017). We then identified the number of DEGs that were also found to be under selection from whole-genome resequencing. A Fisher’s exact test was run to determine if genes within identified significant windows were enriched in DEGs.
Sequence Divergence
To test whether windows under selection also contained genes with high sequence divergence between populations, we compared sequencing reads from the pure Bodega population to the San Diego and Bodega reference genome. Using snpEff, a tool to annotate and predict the effects of SNPs (Cingolani et al. 2012), we annotated the VCF file created from the pure Bodega population mapped to the San Diego reference. We then used a python script to identify Ka/Ks ratios for each gene using the annotated VCF file (Cadzow et al. 2014). The results of a Student’s t-test for the observed P value was compared with the distribution of 10,000 permutations to determine if genes within identified significant windows also had a divergent Ka/Ks ratio from the genome-wide mean Ka/Ks ratio.
Acknowledgments
This work was supported by a Sloan Foundation Grant and a Louisiana Board of Regents Grant awarded to M.W.K. Sequencing data were analyzed using high-performance computing resources provided by Louisiana State University (http://www.hpc.lsu.edu, last accessed November 15, 2020).
Data Availability
The LT50 data and the R and bash scripts are available at https://github.com/JoannaGriffiths/Tigriopus_HER. Raw sequencing data are deposited in the National Center for Biotechnology Information’s Short Reads Archive (accession no.: PRJNA597336).