 Group II Introns Generate Functional Chimeric Relaxase Enzymes with Modified Specificities through Exon Shuffling at Both the RNA and DNA Level
Group II Introns Generate Functional Chimeric Relaxase Enzymes with Modified Specificities through Exon Shuffling at Both the RNA and DNA Level
Molecular Biology and Evolution


- Altmetric
Group II introns are large self-splicing RNA enzymes with a broad but somewhat irregular phylogenetic distribution. These ancient retromobile elements are the proposed ancestors of approximately half the human genome, including the abundant spliceosomal introns and non-long terminal repeat retrotransposons. In contrast to their eukaryotic derivatives, bacterial group II introns have largely been considered as harmful selfish mobile retroelements that parasitize the genome of their host. As a challenge to this view, we recently uncovered a new intergenic trans-splicing pathway that generates an assortment of mRNA chimeras. The ability of group II introns to combine disparate mRNA fragments was proposed to increase the genetic diversity of the bacterial host by shuffling coding sequences. Here, we show that the Ll.LtrB and Ef.PcfG group II introns from Lactococcus lactis and Enterococcus faecalis respectively can both use the intergenic trans-splicing pathway to catalyze the formation of chimeric relaxase mRNAs and functional proteins. We demonstrated that some of these compound relaxase enzymes yield gain-of-function phenotypes, being significantly more efficient than their precursor wild-type enzymes at supporting bacterial conjugation. We also found that relaxase enzymes with shuffled functional domains are produced in biologically relevant settings under natural expression levels. Finally, we uncovered examples of lactococcal chimeric relaxase genes with junctions exactly at the intron insertion site. Overall, our work demonstrates that the genetic diversity generated by group II introns, at the RNA level by intergenic trans-splicing and at the DNA level by recombination, can yield new functional enzymes with shuffled exons, which can lead to gain-of-function phenotypes.
Introduction
Group II introns are large RNA enzymes that are widely found throughout bacteria, some archaea, and the organelles of certain eukaryotes (Lambowitz and Zimmerly 2011; McNeil et al. 2016). Following transcription, these versatile ribozymes associate with their intron-encoded protein to fold into a catalytically competent 3D RNA structure that concurrently enables ligation of the flanking exons and self-splicing of the intron through different pathways (Fedorova and Zingler 2007; Pyle 2016). The most studied self-splicing pathway is branching, where the intron uses a bulged adenosine residue, located near its 3′ end, as a branchpoint to excise as a lariat. Once released as a lariat, the intron can use the reverse branching pathway to invade target sites in DNA or RNA substrates. Following insertion into DNA substrates, group II introns can function as retromobile elements to form stable DNA copies of themselves by completing either the retrohoming or the retrotransposition mobility pathways (Cousineau et al. 1998; Cousineau et al. 2000; Ichiyanagi et al. 2002). Alternatively, bacterial group II introns can initiate self-splicing through less characterized pathways such as circularization, where they catalyze a trans-splicing reaction by recruiting an external nucleophile (Monat et al. 2015; Monat and Cousineau 2016).
When seen through an evolutionary lens, group II introns are believed to have played a monumental role in the evolution of eukaryotes. Due to conserved facets of their biochemical and structural properties, group II introns are the proposed evolutionary ancestors of spliceosomal introns and the non-long terminal repeat retrotransposons, which together account for over half of the human genome (Lambowitz and Belfort 2015; McNeil et al. 2016). Although spliceosomal introns are largely seen as beneficial to eukaryotes by increasing their genetic diversity and overall complexity through regulated pathways such as alternative splicing and trans-splicing (Irimia and Roy 2014; Bush et al. 2017), an opposite view has emerged for their bacterial ancestors. Bacterial group II introns are indeed considered as detrimental, selfish elements subjected to negative selection. They are thought to invade other DNA target sites in order to spread and survive, using splicing solely as a means of preventing damage to their hosts (Dai and Zimmerly 2002; Leclercq and Cordaux 2012). Although ancestral group II introns were most likely selfish and proliferative before the emergence of the first eukaryotes, their current descendants found in various prokaryotic genomes may have evolved specific benefits to their hosts. In this study, we challenge the current paradigm that modern bacterial group II introns behave exclusively as selfish retroelements.
We recently characterized a new group II intron intergenic trans-splicing pathway that increases the genetic diversity of its bacterial host at the RNA level by combining aspects of both the branching and the circularization pathways (LaRoche-Johnston et al. 2018). In this novel splicing pathway, excised group II intron lariats first recognize sequence motifs on bacterial mRNA substrates through base pairing and invade them via reversal of the branching pathway. The intron can target multiple sites on mRNAs since the 11-nt interaction can occur with some mismatches and also because most of the intron sequence motifs (10/11) are made of Gs and Us that can potentially base pair with Cs or Us and As or Gs, respectively. Once inserted in a noncognate host mRNA, the intron excises using the circularization pathway, where it trans-splices an external RNA to its downstream mRNA fragment, thus forming a chimeric mRNA (LaRoche-Johnston et al. 2018). The stochastic nature of this pathway should most of the time result in mRNA–mRNA chimeras that would code for nonfunctional proteins. Although we demonstrated the production of a variety of E1–mRNA and mRNA–mRNA chimeras in vivo, it remained unclear whether these compound transcripts are functional and biologically relevant. We thus examined the native biological context of the model group II intron Ll.LtrB from the gram-positive bacterium Lactococcus lactis to determine whether group II intron-generated chimeric mRNAs are translated and if their corresponding chimeric proteins are functional.
Ll.LtrB interrupts the ltrB gene, which codes for a conjugative relaxase enzyme, LtrB. This single-strand endonuclease is part of a DNA processing complex called relaxosome that assembles at the origin of transfer (oriT) of conjugative elements (Smillie et al. 2010). A key partner of LtrB in the lactococcal relaxosome is the MobC-family accessory protein LtrF, which binds an inverted repeat directly adjacent to the oriT. This interaction is essential for the specific recruitment of LtrB to the oriT (Chen et al. 2007). Once the LtrB–LtrF–oriT complex is formed, the dsDNA is locally unwound, allowing a direct interaction between LtrB and the oriT–ssDNA (Chen et al. 2007). Once bound to ssDNA, LtrB initiates conjugation by nicking oriT and remains covalently bound to the liberated 5′-phosphate (Byrd and Matson 1997). Through protein–protein interactions, the relaxosome next binds to a type 4 coupling protein (T4CP) ATPase at the host membrane, which directs the conjugative element through a mating channel formed by a type 4 secretion system (T4SS) (Chen et al. 2008). By interrupting relaxase genes of various conjugative elements, group II introns use conjugation as a means of survival by spreading to different bacterial strains and species (Belhocine et al. 2004, 2005; Belhocine, Mandilaras, et al. 2007). Moreover, since the relaxase enzyme is an essential component of the conjugative machinery, the accuracy and efficiency of Ll.LtrB self-splicing from the mRNA essentially controls the conjugation of its host element (Mills et al. 1996; Klein et al. 2004). LtrB was also shown to have off-target DNA nicking activity that stimulates both the frequency and diversity of Ll.LtrB retrotransposition events, revealing yet another link between group II intron dissemination and bacterial conjugation (Novikova et al. 2014).
Ll.LtrB interrupts a specific site of ltrB, consisting of a highly conserved catalytic histidine triad in the IncP family of relaxases (Pansegrau et al. 1994). The conserved nature of this catalytic motif has been proposed to be advantageous for the dissemination of Ll.LtrB in L. lactis, providing an abundance of unoccupied sites to invade in orthologous relaxase genes (Staddon et al. 2004). Indeed, we previously described a recent burst of mobility by Ll.LtrB variants within various L. lactis strains and subspecies, nearly all of which specifically invaded the conserved histidine triad (LaRoche-Johnston et al. 2016). We thus hypothesized that the conserved nature of the relaxase recognition site and the frequent exposure of introns to orthologous relaxase genes may enable the consistent production of chimeric relaxase mRNAs containing shuffled exons, which could then be translated into chimeric enzymes with potentially altered functions.
Here, we demonstrate that one of the effects of group II introns increasing bacterial genetic diversity is the production of chimeric relaxase mRNAs and active enzymes under biologically relevant conditions. We show that since relaxase exons exert different functions during conjugation, group II intron trans-splicing of exons from orthologous relaxase mRNAs can produce chimeric enzymes with gain-of-function phenotypes that enhance the spread of conjugative elements. Finally, we also uncovered examples of chimeric relaxase genes throughout L. lactis strains and subspecies with junctions located precisely at the site of group II intron insertion. Overall, our data show for the first time that group II introns can be beneficial to their hosts by producing novel compound transcripts and proteins of functional value to the bacteria, which may have played an important role in the rapid adaptation of L. lactis to the dairy environment.
Results
The Ll.LtrB Group II Intron from L. lactis Generates mRNA and Protein Chimeras between Orthologous Relaxase Genes In Vivo
We previously demonstrated that the Ll.LtrB group II intron mediates the formation of various E1–mRNA and mRNA–mRNA chimeras in L. lactis through a novel intergenic trans-splicing pathway (LaRoche-Johnston et al. 2018). To assess whether Ll.LtrB could use this pathway to generate in-frame chimeric relaxase mRNAs that are functional substrates for translation, we built a group II intron trans-splicing assay containing the relaxase genes from L. lactis and Enterococcus faecalis (fig. 1). In their native environment, the highly similar and homologous Ll.LtrB and Ef.PcfG group II introns interrupt respectively the ltrB and pcfG orthologous relaxase genes at the exact same conserved position at the junction between two codons (LaRoche-Johnston et al. 2016) (fig. 1A). As a consequence of the recent lateral transfer of Ef.PcfG from E. faecalis to L. lactis (LaRoche-Johnston et al. 2016), the flanking exons of the relaxase genes are significantly less conserved (E1: 60%, E2: 58%) than the introns they harbor (99.7%) (fig. 1A).

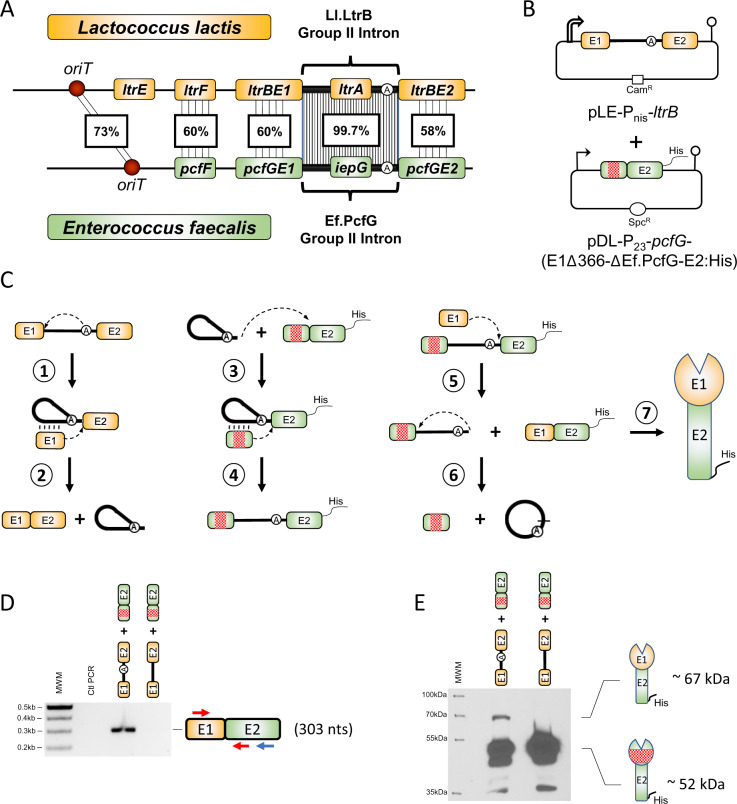
The Ll.LtrB group II intron generates chimeric relaxase mRNAs and proteins in Lactococcus lactis. (A) Comparison of the genetic loci involved in conjugative transfer of the L. lactis pRS01 plasmid (orange) harboring the Ll.LtrB group II intron and the Enterococcus faecalis pTEF4 plasmid (green) harboring the Ef.PcfG group II intron. Both introns interrupt a conserved catalytic motif at the exact same position in the ltrB and pcfG orthologous relaxase genes. Similarities between orthologous genes are shown as percent nucleotide identity. oriT (red circle): origin of conjugative transfer. (B) Two-plasmid system (pLE and pDL) used to study the production of chimeric relaxase mRNAs and proteins in L. lactis. The ltrB relaxase gene (orange) is expressed from a nisin-inducible promoter (open broken arrow) and is interrupted by the Ll.LtrB group II intron, whereas the pcfG relaxase gene (green) is expressed from a constitutive P23 promoter (broken arrow). The pcfG gene also harbors a C-terminal 6× His-tag and a 366-nt in-frame deletion in E1 (red square). (C) Group II intron intergenic trans-splicing pathway producing chimeric relaxases in vivo (LaRoche-Johnston et al. 2018). When Ll.LtrB excises through the branching pathway from the ltrB pre-mRNA (orange), the bulged adenosine’s 2′ OH attacks the 5′ splice site, forming a branched lariat still attached to exon 2 (E2), whereas exon 1 (E1) remains associated with the intron solely through base pairing interactions (vertical lines) (step 1). In the second step of branching, the 3′ OH of the last nt of released E1 attacks the 3′ splice site, ligating both exons and releasing the intron lariat (step 2). Ll.LtrB intron lariats can base pair with a sequence coding for conserved catalytic residues in the non-interrupted orthologous pcfG mRNA (green) and invade it by complete reverse splicing (steps 3 and 4). Introns that interrupt the pcfG mRNA can self-splice using the circularization pathway by recruiting free ltrBE1 (orange) to attack the 3′ splice site, producing a chimeric relaxase mRNA (ltrBE1–pcfGE2, orange–green) marked with a 6× His-tag (step 5). The 2′ OH of the intron’s last nucleotide then attacks the 5′ splice site, generating a head-to-tail intron circle and free E1 (step 6). The chimeric relaxase mRNA (orange–green) can be translated into a His-tagged chimeric relaxase enzyme (orange–green) (step 7). The two-plasmid expression system shown in (B) was used to screen for the in vivo production of chimeric relaxases at the mRNA (D) and protein (E) levels where the ltrB gene was interrupted by either the Ll.LtrB-WT or the Ll.LtrB-ΔA intron. Arrows denote the relative position of primers used for RT-PCR (blue for RT, red for PCR) and the expected size for mRNA chimeras containing a perfect ltrBE1–pcfGE2 junction is shown (303 nt). Expected sizes of translated chimeric (∼67 kDa) and contiguous (∼52 kDa) relaxase proteins are also shown.
The group II intron trans-splicing assay consisted of coexpressing the ltrB and pcfG relaxase genes in L. lactis from the pLE and pDL plasmids, respectively (fig. 1B). The ltrB gene was under the control of the nisin-inducible promoter (Pnis) and interrupted by its cognate Ll.LtrB intron, whereas the non-interrupted pcfG gene was expressed from a constitutive promoter (P23) (fig. 1B). The previously described group II intron intergenic trans-splicing pathway (fig. 1C) (LaRoche-Johnston et al. 2018) predicts that free E1 from the interrupted mRNA (ltrBE1) should be trans-spliced to E2 of the non-interrupted mRNA (pcfGE2), precisely at the E1–E2 splice junction (fig. 1C, step 5). We thus performed a reverse transcriptase-polymerase chain reaction (RT-PCR) assay across the predicted chimeric ltrBE1–pcfGE2 mRNA splice junction from total RNA extracts of L. lactis expressing the ltrB and pcfG genes (Monat et al. 2015; Monat and Cousineau 2016; LaRoche-Johnston et al. 2018). An amplicon of the expected size was obtained specifically when ltrB was interrupted by Ll.LtrB-WT and was absent for the branchpoint mutant, Ll.LtrB-ΔA (fig. 1D). The Ll.LtrB-ΔA negative control cannot support E1 trans-splicing since it is unable to splice through the requisite branching (fig. 1C, steps 1 and 2) and reverse branching pathways (fig. 1C, steps 3 and 4) (Monat et al. 2015; Monat and Cousineau 2016). Sequencing of the RT-PCR amplicon confirmed the identity of the ltrBE1–pcfGE2 chimeric mRNA where E1 of the interrupted ltrB mRNA was precisely ligated to E2 of the pcfG non-interrupted relaxase transcript.
To analyze whether the chimeric ltrBE1–pcfGE2 relaxase transcript is translated into a chimeric protein (fig. 1C, step 7), the non-interrupted pcfG gene included a 6× His-tag at the C-terminus as well as an in-frame deletion of 366 nt in E1 (fig. 1B). When we performed Western blots using His-tag primary antibodies on total protein extracts, a strong signal at ∼52 kDa was detected when ltrB was interrupted by either Ll.LtrB-WT or Ll.LtrB-ΔA (fig. 1E). This band corresponds to the contiguous PcfG relaxase protein harboring a deletion in E1 (PcfGE1Δ366–PcfGE2). We also observed an additional band at ∼67 kDa that corresponds to the size of the chimeric LtrBE1–PcfGE2 relaxase protein exclusively when ltrB was interrupted by Ll.LtrB-WT (fig. 1E).
Overall, our data show that the Ll.LtrB group II intron can catalyze the formation of in-frame chimeric relaxase transcripts as well as detectable levels of chimeric relaxase proteins in L. lactis.
Chimeric Relaxase Enzymes Are Active in L. lactis and Can Confer a Gain-of-Function Phenotype
Having demonstrated that Ll.LtrB can generate chimeric relaxase proteins in L. lactis, we next wanted to assess whether these enzymes were active. We thus engineered a conjugation assay to study the activity of chimeric relaxase enzymes using NZ9800ΔltrB and LM0231 as the donor and recipient strain, respectively (fig. 2A) (Belhocine et al. 2004). The donor strain contained all the conjugation machinery to support the transfer of conjugative elements harboring an origin of transfer (oriT), except for the ltrB relaxase gene. Donor cells were cotransformed with two plasmids, one expressing wild-type or chimeric relaxase enzymes, whereas the second plasmid harbored an L. lactis or an E. faecalis oriT (fig. 2A). This conjugation assay allowed us to study the efficiency with which different relaxase enzymes recognize various oriTs and support the transfer of mobilizable plasmids between strains of L. lactis (fig. 2B).
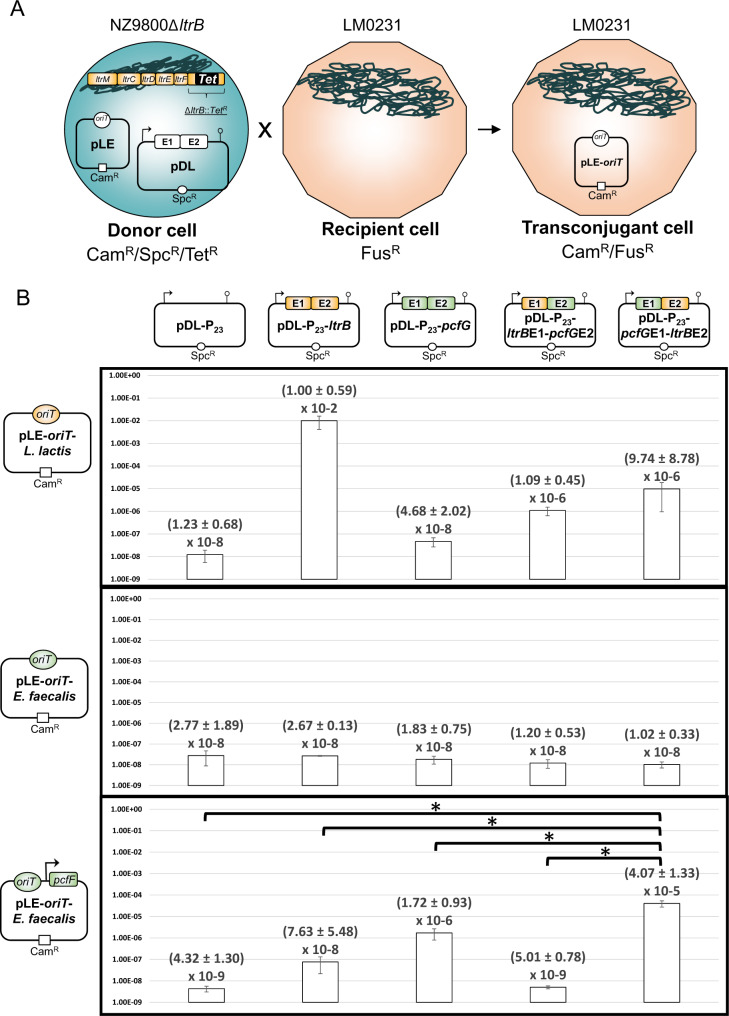
Conjugation efficiencies of plasmids harboring various oriTs in the presence of wild-type or chimeric relaxase enzymes. (A) Schematic representation of the conjugative assay. Lactococcus lactis donor cells (NZ9800ΔltrB) encode the required lactococcal transfer machinery (Ltr genes, orange) to fully support conjugation, except for the ltrB relaxase gene which was replaced by a gene conferring resistance to tetracycline (TetR). Different relaxase genes were supplied in trans from a pDL-based plasmid (SpcR) using a P23 constitutive promoter in the presence of a pLE-based plasmid (CamR) harboring either the L. lactis pRS01 (orange) or the Enterococcus faecalis pTEF4 (green) oriT. Lactococcus lactis recipient cells (LM0231) are resistant to fusidic acid (FusR), and transconjugant cells were isolated by selecting for recipient cells that received the oriT-containing plasmid (CamR/FusR). (B) Conjugation efficiencies for wild-type (LtrB, orange–orange and PcfG, green–green) and chimeric (LtrBE1–PcfGE2, orange–green and PcfGE1–LtrBE2, green–orange) relaxases. A pDL-based plasmid without any relaxase was used to determine background levels of conjugation. Conjugation efficiency was calculated by dividing the number of transconjugant cells by the number of donor cells, which is shown together with the standard error. Each data point represents triplicates of independent assays. Bent arrows denote the presence of a P23 constitutive promoter. Asterisks denote statistical significance (*P < 0.05).
We first looked at the ability of four different relaxase enzymes to support the transfer of a plasmid harboring the oriT from the L. lactis pRS01 plasmid (Mills et al. 1996) (fig. 2B, first row). As expected, the cognate LtrB relaxase from L. lactis supported conjugative transfer of the mobilizable plasmid very efficiently at 106-fold over background levels. However, the PcfG relaxase from E. faecalis was not able to support the transfer of the L. lactis oriT-containing plasmid, showing a conjugation efficiency at background levels. However, when we tested both permutations of chimeric relaxases, we found that LtrBE1–PcfGE2 (88-fold) and PcfGE1–LtrBE2 (791-fold) each supported conjugation efficiencies at low levels but nevertheless clearly above background. These data demonstrate that both chimeric relaxase enzymes are translated, fold appropriately, and can actively support the transfer of an L. lactis-containing oriT plasmid, albeit at lower levels than the cognate wild-type LtrB relaxase.
In contrast, none of the four relaxases was able to support the transfer of a plasmid containing an oriT from the E. faecalis pTEF4 plasmid (LaRoche-Johnston et al. 2016), including its cognate wild-type PcfG relaxase (fig. 2B, second row). These data suggest a functional uncoupling between the oriT–PcfG relaxase unit from E. faecalis and the lactococcal conjugation machinery (ltr genes) encoded by the L. lactis chromosomal sex factor. These two functional units are most likely unable to interact and successfully mediate conjugation of the mobilizable plasmid.
We next assessed the conjugation efficiency of a third mobilizable plasmid that harbored both the E. faecalis oriT and pcfF accessory gene (fig. 2B, third row). PcfF plays an essential role in E. faecalis conjugation, since its deletion results in the complete shutdown of conjugation (Chen et al. 2007). This MobC-family accessory protein is believed to act the same way as the L. lactis orthologous LtrF protein, by binding to the palindromic sequence directly adjacent to the oriT and recruiting the PcfG relaxase to initiate conjugation (Staddon et al. 2006). This construct revealed a ∼400-fold increase in conjugation above background level for the wild-type PcfG relaxase which under biological conditions interacts with the E. faecalis oriT–PcfF complex (fig. 2B, third row). A smaller ∼18-fold increase in conjugation was also detected for the wild-type LtrB relaxase suggesting that there is a limited ability of the lactococcal relaxase to recognize and interact with the E. faecalis oriT–PcfF complex. However, the PcfGE1–LtrBE2 chimeric relaxase showed the largest significant increase in conjugation over background (∼9,420-fold) as well as a significant increase when compared with both wild-type LtrB (∼530-fold) and PcfG (∼24-fold) enzymes. In sharp contrast, the LtrBE1–PcfGE2 chimeric relaxase was not able to support any level of conjugation.
Taken together, our results demonstrate that chimeric relaxase enzymes are active in vivo and can lead to a gain-of-function phenotype when E1 and E2 are associated respectively with their cognate oriT and conjugation machinery. These results also suggest that the minimal components required by a chimeric relaxase enzyme to support the conjugative transfer of a mobilizable plasmid are its cognate oriT and MobC-family accessory protein.
An Ef.PcfG-Generated Chimeric Relaxase Enzyme Supports Conjugation in L. lactis
We next engineered a conjugation assay in L. lactis to determine whether chimeric relaxase enzymes produced as a result of group II intron catalysis could also be active and sustain conjugation (fig. 3). Since the highest conjugation efficiency was detected when both the oriT and pcfF from E. faecalis were coupled with the PcfGE1–LtrBE2 chimeric relaxase (fig. 2B, third row), we cotransformed a pLE plasmid containing the E. faecalis oriT, the pcfF accessory protein, and the Ef.PcfG-interrupted pcfG relaxase with a pDL-based plasmid expressing the non-interrupted ltrB gene (fig. 3A). To reduce background levels of conjugation stemming from both wild-type relaxases, we introduced in-frame deletions of 360 nt in ltrBE1 and 936 nt in pcfGE2 (fig. 3, red boxes). By inactivating both wild-type relaxases, the only remaining way for the pLE plasmid to be transferred by conjugation from NZ9800ΔltrB to LM0231 is by the generation of an intergenic PcfGE1–LtrBE2 relaxase chimera that can functionally bridge the gap between the E. faecalis oriT and the L. lactis conjugation machinery.
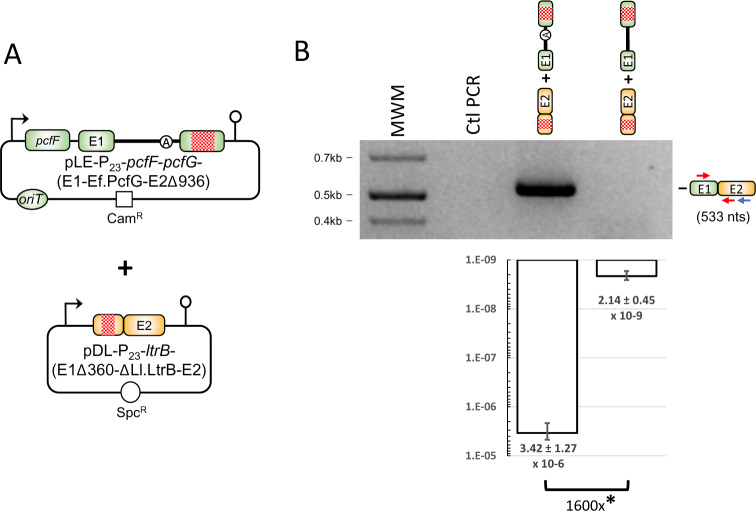
Ef.PcfG-generated chimeric relaxase enzyme supports conjugation in Lactococcus lactis. (A) Conjugation system used to study the production of chimeric PcfGE1–LtrBE2 relaxase enzyme in vivo. The pDL and pLE plasmids are cotransformed in the NZ9800ΔltrB donor strain, whereas the L. lactis strain LM0231 is used as the recipient. Red boxes denote in-frame deletions in pcfGE2 (936 nt) and ltrBE1 (360 nt). Broken arrows represent the P23 constitutive promoter. (B) The production of chimeric pcfGE1–ltrBE2 relaxase mRNA was assessed with the ltrB gene interrupted by either the Ef.PcfG-WT or the Ef.PcfG-ΔA intron. Arrows denote the relative position of primers used for RT-PCR (blue for RT, red for PCR), and the expected size for the mRNA chimera containing a perfect pcfGE1-ltrBE2 junction is shown (533 nt). Conjugation efficiencies were measured as the ability of L. lactis to transfer the pLE-P23-pcfF-pcfG-(E1-Ef.PcfG-E2Δ936) plasmid harboring either the Ef.PcfG-WT or the Ef.PcfG-ΔA intron from the donor to the recipient strain. Conjugation efficiency was calculated by dividing the number of transconjugant cells by the number of donor cells and shown with the standard error. Each data point represents triplicates of independent assays. Asterisks denote statistical significance (*P < 0.05).
To validate our system, we first performed an RT-PCR to look for the pcfGE1–ltrBE2 mRNA chimera. An amplicon was exclusively generated when the pcfG gene was interrupted by its Ef.PcfG-WT intron (fig. 3B). When we tested our system functionally, we observed a relatively high conjugation efficiency (3.42 × 10−6) in the presence of Ef.PcfG-WT, which represented a slight 12-fold decrease from when the chimeric non-interrupted relaxase gene was directly expressed (4.07 × 10−5) (fig. 2B, third row). Importantly, the conjugation efficiency when the pcfG gene was interrupted by Ef.PcfG-WT had a significant 1,600-fold increase over the branchpoint mutant control, Ef.PcfG-ΔA, where the intron was unable to generate chimeric relaxase mRNA (fig. 3B).
Our data thus show that when group II introns interrupt relaxase genes, they can produce enough chimeric relaxase enzymes in donor cells to mediate significant levels of conjugation. These results also demonstrate that the Ef.PcfG group II intron from E. faecalis can, similarly to Ll.LtrB, generate chimeric relaxase mRNAs and active chimeric enzymes in L. lactis.
The Formation of Chimeric Relaxase Transcripts Occurs under Biologically Relevant Conditions in E. faecalis
Having shown with our expression vectors that group II introns can catalyze the formation of active chimeric relaxase enzymes in L. lactis, we next wanted to see if they could also be produced in biologically relevant conditions under natural expression systems. We chose E. faecalis as the bacterial host to cotransform pEF1071 from E. faecalis (Balla and Dicks 2005) and pLE12 from L. lactis (Mills et al. 1996), because they both harbor relaxase genes under the control of their natural promoters (fig. 4A). pEF1071 harbors the non-interrupted mobA relaxase gene that was previously shown to be invaded by Ef.PcfG in E. faecalis (LaRoche-Johnston et al. 2016). pLE12 contains the Ll.LtrB-interrupted ltrB relaxase gene that stems from the pRS01 L. lactis plasmid (Mills et al. 1996). This plasmid was previously shown to transfer laterally from L. lactis to the JH2-2 lab strain of E. faecalis by conjugation, where it can efficiently replicate and produce active group II intron ribonucleoprotein particles (RNPs) (Belhocine et al. 2004). These plasmids were cotransformed in JH2-2 and maintained using their natural origin of replication. Cotransformants contained both plasmids with no apparent additional bands that typically appear when mobility products are generated due to intron mobility into non-interrupted relaxase genes (fig. 4A, bottom panel) (Cousineau et al. 1998; Belhocine et al. 2004). When intron mobility was analyzed by colony hybridization (Belhocine et al. 2004; Plante and Cousineau 2006), it was found to be very limited with on average 5% of mobA genes interrupted by Ll.LtrB (fig. 4A). Nevertheless, this showed that the ltrB gene is expressed at low levels from its natural promoter in E. faecalis, producing relatively small amounts of active Ll.LtrB RNPs.
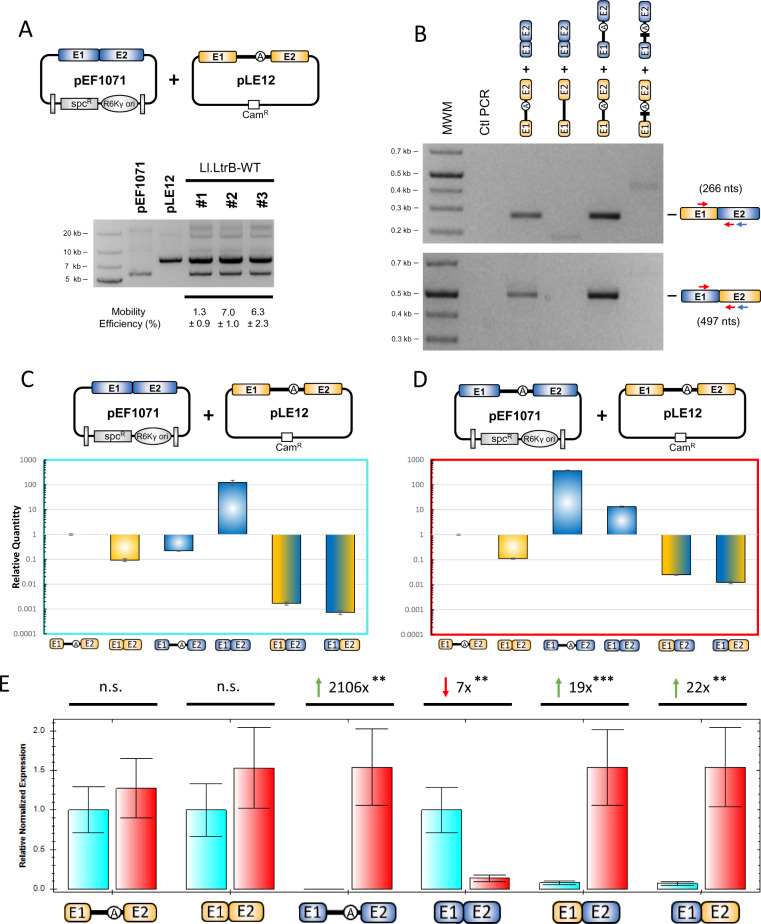
Production of chimeric relaxase mRNAs by Ll.LtrB in biologically relevant conditions in Enterococcus faecalis. (A) The pEF1071 and pLE12 plasmids were cotransformed in the E. faecalis lab strain JH2-2. The pEF1071 plasmid expressed the E. faecalis non-interrupted relaxase mobA gene (blue), whereas the pLE12 plasmid expressed the Lactococcus lactis relaxase ltrB gene (orange) interrupted by the Ll.LtrB group II intron. Both genes were under the control of their natural promoters. Plasmid preparations from three independent cotransformants were run on an agarose gel and shown to contain both plasmids. Mobility efficiency of Ll.LtrB from pLE12 to pEF1071 was calculated through patch hybridization (Plante and Cousineau 2006) and is shown with the standard error. (B) Formation of relaxase mRNA chimeras in different biological settings. RT-PCR was performed to determine the presence of chimeric relaxase mRNAs in JH2-2 strains containing different plasmid combinations. Plasmid combinations from left to right: pLE12 expressing ltrB interrupted by the Ll.LtrB-WT intron with pEF1071 expressing non-interrupted mobA; pLE12 expressing ltrB interrupted by the Ll.LtrB-ΔA intron with pEF1071 expressing non-interrupted mobA; pLE12 expressing ltrB interrupted by the Ll.LtrB-WT intron with pEF1071 expressing mobA also interrupted by the Ll.LtrB-WT intron; pLE12 expressing ltrB interrupted by the Ll.LtrB-ΔDV intron with pEF1071 expressing mobA also interrupted by the Ll.LtrB-ΔDV intron. Arrows denote the relative position of the RT-PCR primers (blue for RT, red for PCR). The relative amounts of various transcripts were compared with the ltrB pre-mRNA precursor by RT-qPCR, in conditions where one (C) or both (D) relaxase genes were interrupted by Ll.LtrB-WT. (E) RT-qPCR of samples producing relaxase chimeras (blue, Ll.LtrB-WT in pLE12; red, Ll.LtrB-WT in both pLE12 and pEF1071). ΔΔCt values were calculated to determine fold change between conditions where either one (blue) or two (red) Ll.LtrB-WT introns were present. Each data point represents technical duplicates done for biological triplicates. Biological replicates were normalized to the ldhB housekeeping gene of E. faecalis, which was used as a reference gene. Green arrows represent increases of a specific amplicon when two group II introns are present, whereas the red arrow represents a decrease. Asterisks denote statistical significance for an unpaired Student’s t-test (**P < 0.01 and ***P < 0.001).
We next addressed qualitative aspects of the mechanism of chimera formation in vivo (fig. 4B). We first found that the natural expression levels of both relaxase genes are sufficient to generate chimeric mRNAs, which is again dependent on the branching pathway since no chimeras are detected when ltrB is interrupted by the branchpoint mutant (fig. 4B, top panel, lanes 3–4). Surprisingly, although our model predicted exclusively the production of ltrBE1–mobAE2 chimeras (fig. 1C), we also detected the presence of counterpart chimeras, mobAE1–ltrBE2 (fig. 4B, bottom panel, lanes 3–4). A potential explanation is that expression of the Ll.LtrB-interrupted mobA gene, resulting from the mobility of Ll.LtrB from pLE12 to pEF1071, leads to the production of free mobAE1 (fig. 1C, step 1) and the unexpected mobAE1–ltrBE2 chimeras (fig. 1C, step 5). We thus next modified our system to simulate a 100% mobility scenario, where both relaxase genes are fully interrupted by Ll.LtrB. We detected stronger RT-PCR amplicons for both mRNA chimeras, suggesting a positive correlation between intron invasion of a target site and mRNA chimera formation (fig. 4B, both panels, lane 5). Finally, to determine whether mRNA chimera formation is a product of group II intron catalysis or some type of RNA and/or DNA recombination event, we modified our assay so that both introns lacked their small catalytic domains (Ll.LtrB-ΔDV) (Zhao and Pyle 2017). Ll.LtrB-ΔDV is completely inactive and unable to perform any type of splicing reaction. However, the small deletion left the bulk of the intron sequences intact (2,459/2,492 nt) while maintaining perfect sequence homology between both intron copies. Interestingly, neither type of chimeric mRNAs were detected when both relaxase genes are interrupted by Ll.LtrB-ΔDV (fig. 4B, both panels, lane 6).
We next used our assays with one or two wild-type introns to quantitatively address mRNA chimera production by quantitative RT-PCR (RT-qPCR). We began by assessing the relative amounts of various RNAs being produced for each construct, using the ltrB pre-mRNA as the reference. When only one intron is present, interrupting ltrB (fig. 4C), we detected about ten times more ltrB pre-mRNA than ligated exons, whereas the ltrBE1-mobAE2 and mobAE1-ltrBE2 chimeras were respectively 55 and 126 times less abundant than ligated exons. When the two relaxase genes were interrupted by Ll.LtrB (fig. 4D), the proportion of mRNA chimeras appeared to increase. Although splicing efficiency was similar with about nine times more ltrB pre-mRNA than ligated exons, there were now respectively only about four and nine times fewer ltrBE1–mobAE2 and mobAE1–ltrBE2 chimeras than ligated exons. We finally compared the two systems by analyzing the relative normalized expression of each target (fig. 4E). We first found that, as expected, amounts of ltrB pre-mRNA and ligated exons were not significantly different. However, the expression system with two interrupted genes (fig. 4E, red bars) showed significant 19- and 22-fold increases in production of ltrBE1–mobAE2 and mobAE1–ltrBE2 chimeras respectively when compared with the expression system with a single intron (fig. 4E, blue bars), again supporting a positive correlation between intron invasion of a target site and mRNA chimera formation.
Taken as a whole, these results show that chimeric transcripts are produced at detectable levels by Ll.LtrB when the two relaxase genes are present on biologically relevant vectors and expressed under the control of their natural promoters in E. faecalis. Furthermore, our results demonstrate that chimeric transcript formation is dependent on intron catalysis and increases when more target sites are occupied by group II introns.
Phylogenetic Analyses Unveil Group II Intron-Generated Chimeric Relaxase Genes
The Ll.LtrB intron from the lactococcal pRS01 plasmid has been a model system to study group II intron splicing, mobility, lateral transfer as well as evolution. However, at least 60 closely related full-length intron variants are present in different species, subspecies, and strains of lactococci (Candales et al. 2012). These group II introns have over 95% identity to Ll.LtrB and they mostly interrupt orthologous relaxase genes at the exact same conserved position, suggesting a recent acquisition and dissemination into this group of lactic acid bacteria (LaRoche-Johnston et al. 2016). Since most of these introns interrupt relaxase genes, we wanted to study the phylogenetic relationship between the lactococcal relaxase genes that are interrupted by group II introns.
We first analyzed the flanking exon sequences of each intron and found that 53/60 were interrupting genes that could be identified as coding for relaxase enzymes. All intron-containing relaxase genes were interrupted at the exact same position within the conserved catalytic histidine triad common to members of the IncP relaxase family (Pansegrau et al. 1994). To avoid redundancy, we further narrowed our analyses to exclude sequences that were identical on both sides of the intron insertion site (±25 nt), leaving only 16/53 relaxase genes. Phylogenetic trees were then generated using either the full-length genes (fig. 5A), E1 (fig. 5B), or E2 (fig. 5C) by maximum likelihood using PhyML (Guindon et al. 2010), with 1,000 bootstraps and the E. faecalis pcfG relaxase gene from the pTEF4 plasmid as the outgroup.
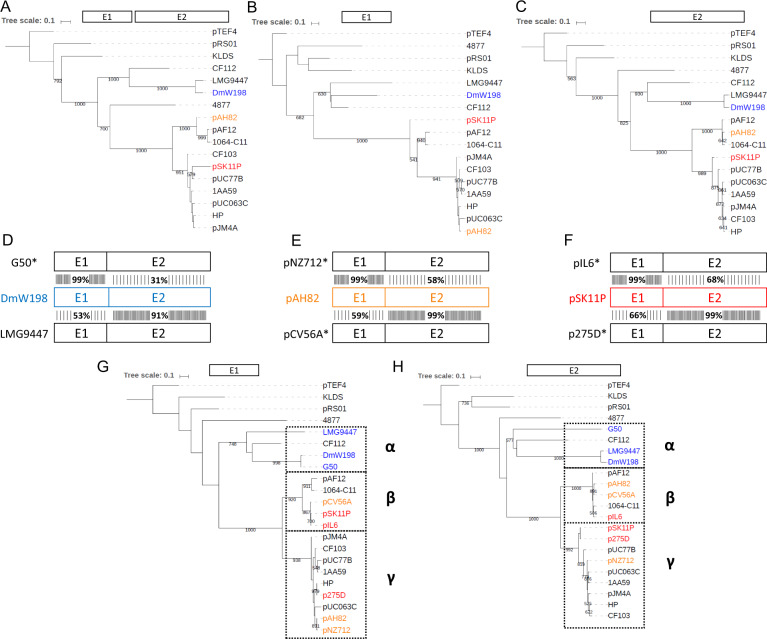
Phylogenetic trees of intron-interrupted relaxase genes from lactococci. The phylogeny of all non-redundant intron-interrupted relaxase genes found in lactococci was assessed by maximum likelihood using PhyML with 1,000 bootstraps. The phylogenetic trees were produced using either the full-length relaxase genes without the group II introns (A), the relaxase sequences preceding the intron (E1) (B), or the relaxase sequences following the intron (E2) (C). In each case, the intronless pcfG relaxase gene from the Enterococcus faecalis pTEF4 plasmid was used as the outgroup. Both exons from the DmW198 (D), pAH82 (E), and pSK11P (F) relaxase genes were individually analyzed by searching for the most similar sequences in GenBank using BlastN. Highest similarity results for E1 are shown above the reference sequence, whereas the highest similarity results for E2 are shown below the reference sequence. Exons of both relaxase genes were then fully compared with the initial query to determine whether homology extended to the entire gene. Relaxase genes with an asterisk denote that they are not interrupted by a group II intron. The relaxase genes marked with an asterisk were included to generate phylogenetic trees with greater resolution for E1 (G) and E2 (H). The α, β, and γ groups delineate separately evolving lineages of relaxase genes in lactococci. Accession numbers of GenBank sequencing projects containing the relaxase genes used are listed in supplementary table S3, Supplementary Material online. Matrices and phylogenetic trees are available in the TreeBASE online repository (URL: http://purl.org/phylo/treebase/phylows/study/TB2:S27000, last accessed November 5, 2020.).
We first noticed that even though the overall structure of the trees was very similar, the position of some sequences was changing drastically between trees. To further investigate the evolutionary relationships of all 16 relaxase genes, we performed BlastN searches using the individual exons of each relaxase genes as an input sequence against the Nucleotide collection database for L. lactis (taxid: 1358). Our goal was to determine whether the relaxase genes in each cluster were distinct monophyletic groups and belonged to the same evolutionary lineage. For each of the 16 E1 and E2 queries, the entire gene of the highest nucleotide identity exon match identified by BlastN was aligned to the whole gene of the input exon to determine whether nucleotide similarity between exons extended to the remainder of the gene. We found three genes which appeared to be made up of exons from different relaxase families: DmW198 (fig. 5D), pAH82 (fig. 5E), and pSK11P (fig. 5F). All three cases have highest similarities with at least one relaxase gene that does not contain a group II intron (fig. 5D–F, asterisks). Interestingly, nucleotide identity steeply drops off for both exons precisely at the intron insertion site, such that each gene is drastically different from the exons that were not specifically used as input sequences (fig. 5D–F).
To increase the resolution of our initial trees, we generated new phylogenetic trees of E1 (fig. 5G) and E2 (fig. 5H) that included the five additional genes found by searching for closest exon matches (fig. 5D–F, asterisks). If relaxase genes had evolved as monophyletic units, we would expect no significant changes between the makeup of phylogenetic trees made for E1 or E2. We found that although the newly added relaxase genes remained in the same distinct clusters of the trees regardless of the exon that was analyzed, the exons of the pAH82 and pSK11P relaxase genes clearly belonged to different clusters. The DmW198 relaxase gene remained in the α cluster for both exons, likely representing a chimera formed between more closely related relaxase genes whose chimeric nature would likely be more obvious if the tree had better resolution.
Overall, these data indicate that certain lactococci contain relaxase genes whose exons have different evolutionary origins, since both exons belong to distinct relaxase phylogenetic lineages. Furthermore, these chimeric relaxase genes were likely generated by group II intron-based exon shuffling, since the point at which nucleotide homology shifts correspond precisely to the site of group II intron insertion.
Discussion
We previously described how reversal of the group II intron branching pathway into ectopic mRNAs produces a population of intron-interrupted cellular transcripts (fig. 1C, steps 3 and 4). When used as templates for circularization instead of branching, these intron-interrupted mRNAs were shown to generate a population of E1–mRNA and mRNA–mRNA chimeric transcripts (fig. 1C, steps 5 and 6) (LaRoche-Johnston et al. 2018). We thus proposed that this new group II intron intergenic trans-splicing pathway increases genetic diversity at the RNA level by shuffling coding sequences with potential benefits to the host cell. However, it remained unclear whether these chimeric transcripts are recognized by ribosomes and translated into chimeric proteins in sufficient amounts to yield any observable phenotype.
In this study, we took advantage of the native biological context of the Ll.LtrB and Ef.PcfG group II introns from L. lactis and E. faecalis to address important features of the intergenic trans-splicing pathway. Ll.LtrB and Ef.PcfG interrupt respectively the ltrB and pcfG orthologous relaxase genes at the same position between two codons (fig. 1A), potentially allowing for in-frame exon shuffling between their mRNAs through intergenic trans-splicing (fig. 1C) (LaRoche-Johnston et al. 2016).
We first demonstrated that Ll.LtrB can generate ltrBE1–pcfGE2 chimeric transcripts between the interrupted ltrB and non-interrupted pcfG relaxase mRNAs (fig. 1D) and that these mRNA–mRNA chimeras are recognized by the translation machinery leading to the production of detectable amounts of LtrBE1–PcfGE2 chimeric proteins (fig. 1E) in L. lactis.
Next, we showed that chimeric relaxase enzymes between LtrB and PcfG are active in L. lactis and can even confer a gain-of-function phenotype when compared with their precursor wild-type enzymes (fig. 2). Previous in vitro work on the specificity determinants of the L. lactis and E. faecalis conjugative systems had shown that the LtrF accessory protein from L. lactis could functionally substitute the E. faecalis PcfF protein in recognition of the E. faecalis oriT and recruitment of the PcfG relaxase (Chen et al. 2007). However, we found the conjugation efficiency of the PcfG relaxase in our lactococcal system, where LtrF is expressed from the chromosome, to be at background levels (fig. 2B, second row). When the cognate PcfF accessory protein was provided in L. lactis, the transfer efficiencies supported by certain relaxases increased (fig. 2B, third row). Interestingly, even though we detected small increases in conjugation efficiency for the two wild-type relaxase enzymes, the efficiency of the PcfGE1–LtrBE2 relaxase was significantly higher, whereas its counterpart LtrBE1–PcfGE2 was completely inactive. The ability of the PcfGE1–LtrBE2 chimeric relaxase to considerably outperform both wild-type relaxases likely stems from the architecture of these enzymes, which are generally composed of two domains. The N-terminal domain, corresponding to E1, contains three distinct motifs which are believed to act in concert to bind and nick ssDNA at the oriT and to form a covalent bond between the liberated 5′ phosphate of the ssDNA and a highly conserved tyrosine residue (Byrd and Matson 1997). This was supported by functional assays showing that relaxase enzymes with truncated C-terminal ends were sufficient to recognize and nick their cognate oriT and yet were unable to complete conjugative transfer through the mating pore (van Kregten et al. 2009; Cascales et al. 2013). The larger C-terminal domain, corresponding to E2, is less well characterized, having very little sequence conservation (Pansegrau et al. 1994). However, it plays an essential role during conjugation and has recently been shown to bind the all-alpha domain of T4CPs, thus conferring specificity to distinct T4SSs (Whitaker et al. 2015). Taken together, the separation of functional domains of relaxase enzymes thus supports a model where E1 of a chimeric relaxase recognizes, binds, and nicks its cognate oriT, in conjunction with its MobC-family accessory protein, whereas the function of E2 is to provide specificity to the larger T4SS through interactions with the T4CPs. This molecular architecture is consistent with our conjugation data, which showed that the best suited relaxase to interact with the E. faecalis oriT and L. lactis T4CP, PcfGE1–LtrBE2, indeed gave the highest conjugation efficiency. Conversely, the worst-suited relaxase to interact with its binding partners is expected to be the complement chimeric relaxase, LtrBE1–PcfGE2, which was accordingly at background levels and the least efficient of all relaxases tested.
We then determined that a chimeric relaxase enzyme, produced through intergenic trans-splicing in vivo, is abundant enough to yield an observable phenotype. We used an L. lactis conjugation assay where the two precursor relaxase genes harbored large deletions in either E1 or E2, such that conjugation is only detectable when the two functional exons of each relaxase gene are shuffled together (fig. 3A). When the pcfG gene was interrupted by Ef.PcfG-WT we observed high conjugation efficiency, which completely disappeared when the intron was mutated to lack the branchpoint adenosine (fig. 3B). The ability of chimeric relaxases, produced in small amounts when compared with contiguous relaxases, to produce a gain-of-function phenotype may be due to the fact that only a limited amount of relaxase enzyme is necessary to successfully mediate conjugation (Chen et al. 2005; Belhocine, Mak, et al. 2007). Our data also showed that Ef.PcfG from E. faecalis can induce exon shuffling between the ltrB and pcfG mRNAs in L. lactis through intergenic trans-splicing similarly to Ll.LtrB.
We subsequently demonstrated that Ll.LtrB can generate mRNA chimeras under biologically relevant conditions in E. faecalis. Despite the fact that the ltrB and mobA relaxase genes were expressed from their natural promoters, we were able to detect ltrBE1–mobAE2 chimeras by RT-PCR. In accordance with our previous results, this amplicon was absent when ltrB was interrupted by the Ll.LtrB-ΔA branchpoint mutant (LaRoche-Johnston et al. 2018). Unexpectedly, we also detected mobAE1–ltrBE2 chimeras again exclusively for Ll.LtrB-WT. Our model (fig. 1C) predicted a clear directionality for the intergenic trans-splicing pathway, which would favor the sole production of ltrBE1–mobAE2 chimeras. Expression of the Ll.LtrB-interrupted mobA gene from mobility products in pEF1071 seems to contribute to the generation of mobAE1–ltrBE2 through the production of free mobAE1. However, an alternative explanation is that the intron may interact with the non-interrupted mobA transcripts in other ways than reverse splicing. For instance, Ll.LtrB lariats may be hydrolyzing the contiguous exons of non-interrupted mobA transcripts by the spliced exon reopening reaction, also leading to the release of free mobAE1 (Qu et al. 2018).
We next determined the relative abundance of both ltrBE1–mobAE2 and mobAE1–ltrBE2 compared with ltrBE1–ltrBE2 in contexts where ltrB (fig. 4C) or ltrB and mobA (fig. 4D) are interrupted by Ll.LtrB. Despite the fact that internal comparisons could be slightly biased due to the use of different primer pairs for each target (Yuan et al. 2006), chimeric mRNA formation was much higher than we expected. In a biologically relevant context where only ltrB is fully interrupted, in the presence of small amounts of interrupted mobA (Ll.LtrB mobility products) (∼5%) (fig. 4C), both types of chimeras were produced at a proportion of about 1–2% compared with ltrB ligated exons (1.8% for ltrBE1–mobAE2 and 0.8% for mobAE1–ltrBE2). When both relaxase genes were fully interrupted by Ll.LtrB (fig. 4D), this ratio increased by ∼14-fold (25% for ltrBE1–mobAE2 and 11% for mobAE1–ltrBE2). Our findings are further supported by the relative normalized expression analysis between the two systems. When both relaxase genes were interrupted by Ll.LtrB, the two types of chimeras increased by about 20-fold (19-fold for ltrBE1–mobAE2 and 22-fold for mobAE1–ltrBE2) compared with when only ltrB was interrupted (fig. 2E).
The copy number of group II introns is notoriously low within the chromosomes of individual bacteria (Lambowitz and Zimmerly 2011). In addition, group II introns were previously shown to generate recombination events in bacteria when multiple copies were present within the genome (Leclercq et al. 2011). However, using a trans-splicing assay where both relaxase genes are interrupted by catalytically inactive introns (Ll.LtrB-ΔDV), we demonstrated that the mRNA chimeras observed are exclusively produced by intron catalysis and not generated through some type of RNA and/or DNA homologous recombination event (fig. 4B).
On the other hand, by making phylogenetic trees outlining the evolutionary relationships of lactococcal relaxase genes interrupted by a group II intron, we found three genes where the two exons belonged to different evolutionary lineages: DmW198, pAH82, and pSK11P. These chimeric relaxase genes may have been generated at the DNA level through homologous recombination since they are interrupted by almost identical group II introns at the exact same position. Indeed, group II introns and other mobile elements in bacteria such as insertion sequence (IS) elements were previously shown to generate large-scale modifications in bacterial genomes through processes such as recombination (Leclercq et al. 2011). The distinguishing characteristic of chimeric genes generated by group II intron-mediated recombination is their ability to still be expressed as chimeras, due to intron self-splicing at the RNA level, which may limit the potential damage brought on by recombination. If almost identical introns, occupying conserved sites in homologous or orthologous genes, mediate recombination events, the interrupted gene becomes chimeric but may still yield a functional product once the intervening intron splices and ligates its shuffled exons. Alternatively, we cannot completely rule out the possibility that chimeric relaxase genes may have been generated by the reverse transcription of group II intron-generated chimeric mRNAs followed by the fixation of these cDNAs in L. lactis genomes and/or plasmids.
The great majority of bacterial conjugative elements harbor at least the basic components to produce their own relaxosomes consisting of an oriT and oriT-specific relaxase and accessory protein (Smillie et al. 2010). Upon arrival in a new host, a newly transferred non-autonomous mobilizable plasmid is thus in a conjugative cul-de-sac if its relaxase is not recognized by the resident conjugation machinery (fig. 6, scenario 1) or if the relaxase of the resident conjugative element cannot recognize its oriT (fig. 6, scenario 4). However, our data suggest that if at least one of the two relaxase genes, encoded either on the acquired or on the resident conjugative element, is interrupted by a group II intron, then two chimeric relaxase enzymes with shuffled exons can be generated by intergenic trans-splicing (fig. 6, scenarios 2 and 3). One of the two chimeric relaxase enzymes, harboring E1 and E2 specific for the oriT of the mobilizable plasmid and the T4CP encoded by the resident conjugative element, respectively, could bridge the functional gap between the DNA transfer and replication (Dtr) proteins (relaxases and accessory proteins) and the mating pore formation (Mpf) proteins (T4CPs and T4SSs) (fig. 6, scenario 2) of incompatible conjugative systems. This would allow the transfer of the conjugative element even if its own relaxase is unable to do so. Our work also indicates that if the two relaxase genes are interrupted by homologous group II introns, more chimeric relaxases should be produced, in turn supporting higher levels of conjugation. Of importance, the presence of a group II intron-interrupted relaxase gene within the conjugative element of a bacterial host, like for example the L. lactis chromosomal sex factor, should provide a link to its conjugation machinery and stimulate the conjugative transfer of all acquired non-autonomous mobilizable elements.
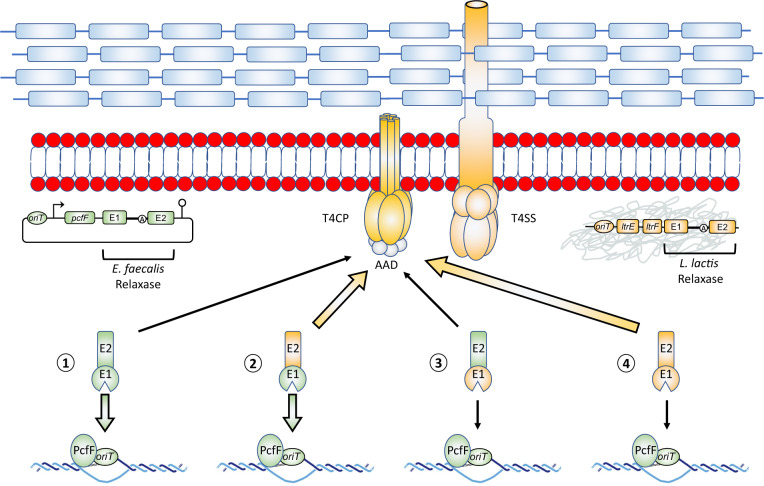
Some group II intron-generated chimeric relaxase enzymes allow the dissemination of conjugative elements by functionally linking incompatible origins of transfer and conjugation machineries. Schematic representation of a bacterial cell harboring a chromosomally embedded conjugative element (orange genes). This conjugative element encodes all the genes required for its own lateral transfer by conjugation: a relaxosome, a T4CP, and a T4SS. When a foreign non-autonomous conjugative element enters the cell, it only contains a minimal set of genes (relaxase, accessory protein) that can form a relaxosome specific for its cognate oriT (green genes). This renders its continued lateral transfer by conjugation contingent on successful interactions with its new host conjugation machinery. The non-autonomous conjugative element is in a conjugative cul-de-sac when the C-terminus of its relaxase (E2) cannot be recognized by the host conjugation machinery (scenario 1) and when the N-terminus of the host relaxase (E1) cannot recognize its oriT (scenario 4). When a group II intron is found interrupting at least one of the relaxase genes, two types of chimeric relaxase enzymes can be generated: green–orange (scenario 2) and orange–green (scenario 3). Conjugative transfer of a non-autonomous conjugative element is most efficient when E1 matches the oriT from the mobilizable plasmid (E1, green), and E2 matches the resident conjugative element (E2, orange) (scenario 2). In this scenario, E1 will interact with its MobC-type accessory protein, and oriT to initiate nicking, whereas E2 will interact with the host T4CP’s all-alpha domain (AAD), which mediates specificity for substrates to be passed along into the T4SS mating pore between the donor and recipient cell during conjugation. The group II intron-generated chimeric relaxase in scenario 2, PcfGE1–LtrBE2, functionally links the Enterococcus faecalis oriT with the Lactococcus lactis conjugation machinery leading to efficient conjugative transfer.
The ability of the Ll.LtrB group II intron to produce functional chimeric relaxases in vivo increases conjugative efficiency, which in turn can affect the relationship of these mobile elements with their bacterial hosts. Group II introns were long thought of as parasitic elements that were solely subjected to negative selection by their bacterial hosts (Leclercq and Cordaux 2012). In the specific case of Ll.LtrB, the interrupted ltrB gene was shown to be translated at lower levels than the non-interrupted ltrB gene (Chen et al. 2005), and even to be targeted for degradation by the group II intron (Qu et al. 2018). However, we show here that group II introns such as Ll.LtrB and Ef.PcfG can in fact be beneficial to their host conjugative elements by catalyzing the formation of chimeric relaxase enzymes that increase their potential of dispersal by conjugation. This appears especially relevant in the biological context of L. lactis where rapid adaptation to the dairy environment largely occurred by shrinking of the bacterial chromosome through reductive evolution and a concurrent drastic increase in plasmid content, most of which were acquired by conjugation (Cavanagh et al. 2015). For L. lactis dairy strains, it is thus likely that the acquisition of new plasmids would have been positively selected for, which may account for the recent dispersal of Ll.LtrB variants that has taken place within different species, subspecies, and strains of dairy lactococci (LaRoche-Johnston et al. 2016).
Taken together, our data show that bacterial group II introns can generate active chimeric relaxase enzymes by shuffling coding sequences at both the RNA level by intergenic trans-splicing and at the DNA level, most likely by homologous recombination. This is the first demonstration that group II introns can be beneficial to the conjugative elements that harbor them and to their bacterial host cells. Although mobilizing into a new DNA site may be seen purely in terms of intron spreading and survival, the ability to increase genetic diversity by generating a new population of mRNA chimeras as well as chimeric genes that could be beneficial to the host cell may be another factor that positively selects for mobility events and eventually fixes them in a population. The specific benefit of the Ll.LtrB variants in lactococci is illustrated by their positive selection in L. lactis, which enabled their recent dissemination in the highly conserved sites of several relaxase genes following the lateral transfer of Ef.PcfG from E. faecalis to L. lactis.
Although the work presented here defies the paradigm that bacterial group II introns provide no benefits to their hosts, it is nevertheless compatible with the fact that these retroelements behave selfishly in order to spread and survive within bacterial cells. Our work thus provides an interesting case study to describe how beneficial outcomes can arise from selfish behavior throughout the course of evolution.
Materials and Methods
Bacterial Strains and Plasmids
Enterococcus faecalis lab strain JH2-2 was grown in Brain Heart Infusion (BHI) media at 37 °C without shaking. Lactococcus lactis strains NZ9800ΔltrB (TetR) (Ichiyanagi et al. 2002) and LM0231 were grown in M17 media supplemented with 0.5% glucose at 30 °C without shaking. Escherichia coli strains DH10β and TransforMax EC100D pir+ were grown in Luria-Bertani (LB) media at 37 °C with shaking. Antibiotics were used at the following concentrations: chloramphenicol (CamR), 10 μg/ml; spectinomycin (SpcR), 300 μg/ml; fusidic acid (FusR), 25 μg/ml. Plasmids used in this study are listed in supplementary table S1, Supplementary Material online. The construction of some plasmids was previously described (pLE-PNis-ltrB [LaRoche-Johnston et al. 2016] and pLE12 [Mills et al. 1996]). The pEF1071 plasmid was isolated from a clinical strain of E. faecalis (SF24397) (McBride et al. 2007). Since this clinical strain is difficult to work with, we performed a Tn5 transposition assay to insert a gene conferring resistance to spectinomycin for selection and the R6Kγori E. coli origin of replication (fig. 4A), generating the pEF1071::<R6Kγori/SpcR> plasmid. Other plasmids were constructed by restriction-digestion/ligation reactions (pDL-P23-pcfG, pDL-P23-ltrB, pLE-oriT-L. lactis, pLE-oriT-E. faecalis, pLE-oriT-E. faecalis-P23-pcfF-pcfG-E2Δ936). The pEF1071::<R6Kγori/SpcR>-Ll.LtrB plasmid was obtained through the invasion of mobA by Ll.LtrB in vivo. The following plasmids were obtained by using the NEB Site-Directed Mutagenesis kit (pLE12-ΔA, pLE12-ΔDV, pEF1071::<R6Kγori/SpcR>-Ll.LtrB-ΔDV, pDL-P23-ltrB-E1Δ360, pLE-P23-pcfF-pcfG-Ef.PcfGΔA-E2Δ936, pDL-P23-pcfG-E1Δ366-ΔEf.PcfG-E2:His). Primers used for site-directed mutagenesis and cloning are shown in supplementary table S2, Supplementary Material online.
RNA Extraction, RT-PCR, and RT-qPCR
Lactococcus lactis cultures were induced with nisin when required and RNA extractions were done on various L. lactis and E. faecalis strains as previously described (Belhocine, Mak, et al. 2007). RT-PCRs for the detection of chimeric mRNAs produced in L. lactis and E. faecalis were done using branchpoint mutant controls and stringent amplification conditions, as previously described (LaRoche-Johnston et al. 2020). RT-qPCR reactions were done by treating total RNA extracts (10 μg/sample) with RNase-free DNase I (New England Biolabs) for 1 h at 37 °C. RNA was then recovered from the reaction (RNeasy Mini Kit, Qiagen). RT reactions were then performed as previously described (Belhocine, Mak, et al. 2007), using an annealing temperature of 50 °C and three RT primers for every target to be analyzed (all primers in supplementary table S2, Supplementary Material online). cDNA was then loaded onto a 96-well PCR plate (Progene), where a qPCR was done using a SYBR-green fluorescent dye (abm) in a qPCR plate reader (Bio-Rad). Results were analyzed using Bio-Rad CFX Manager. Each data point represents the average of technical duplicates done for biological triplicates. No-template controls and no RT controls were also added for each target. The E. faecalis gene Lactate Dehydrogenase B (ldhB) was used as a reference gene across samples for normalization.
Protein Extractions and Western Blotting
Lactococcus lactis cultures were induced with nisin and whole protein extractions were performed as previously described (Hugentobler et al. 2012). Raw protein extracts were run on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (8%), then transferred on a polyvinylidene difluoride (PVDF) membrane for Western blotting as previously described (Hugentobler et al. 2012). 6× His-Tag Monoclonal Antibody (HIS.H8) from Thermo Fisher Scientific (MA1-21315) was used as a primary antibody (1:3,000). Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, conjugated to Horseradish Peroxidase from Thermo Fisher Scientific (G-21040) was used as a secondary antibody (1:5,000).
Mobility and Conjugation Assays
Intron mobility efficiency was calculated by patch hybridization as previously described (Plante and Cousineau 2006), using TransforMax EC100D pir+E. coli cells. Each data point represents triplicates of independent transformants, along with the standard error. Statistical significance was determined using a Student’s unpaired t-test (P < 0.05). Conjugation assays were done on milk plates between L. lactis donor strain NZ9800ΔltrB and L. lactis recipient strain LM0231, as previously described (Belhocine et al. 2004). Each conjugative assay was performed in triplicates, and statistical significance was determined using a Student’s unpaired t-test (P < 0.05).
Phylogenetic Trees
Input sequences were aligned using Clustal Omega (Sievers et al. 2011). Output files in Philip format were then used to generate maximum-likelihood trees in PhyML (Guindon et al. 2010), using nearest neighbor interchange and 1,000 bootstraps. The trees were then visualized using the interactive tree of life software (Letunic and Bork 2011). Accession numbers of GenBank sequencing projects containing the relaxase genes used are listed in supplementary table S3, Supplementary Material online. Matrices and phylogenetic trees are available in the TreeBASE online repository (URL: http://purl.org/phylo/treebase/phylows/study/TB2:S27000).
Data Availability
Some of the data underlying this article are available in the TreeBASE repository and can be accessed at the following link: URL: http://purl.org/phylo/treebase/phylows/study/TB2:S27000.
Acknowledgments
This work was supported by a discovery grant from the Natural Sciences and Engineering Research Council of Canada (227826 to B.C.). F.L.-J. received a Graduate Excellence Fellowship from McGill University, a CGS-M Scholarship from Natural Sciences and Engineering Research Council of Canada, and a Master’s Research Scholarship from Fonds de Recherche en Nature et Technologies and currently holds an Alexander Graham Bell CGS-D Scholarship from Natural Sciences and Engineering Research Council of Canada.