 Evolutionary Genomics of Niche-Specific Adaptation to the Cystic Fibrosis Lung in Pseudomonas aeruginosa
Evolutionary Genomics of Niche-Specific Adaptation to the Cystic Fibrosis Lung in Pseudomonas aeruginosa
Molecular Biology and Evolution


- Altmetric
The comparative genomics of the transition of the opportunistic pathogen Pseudomonas aeruginosa from a free-living environmental strain to one that causes chronic infection in the airways of cystic fibrosis (CF) patients remain poorly studied. Chronic infections are thought to originate from colonization by a single strain sampled from a diverse, globally distributed population, followed by adaptive evolution to the novel, stressful conditions of the CF lung. However, we do not know whether certain clades are more likely to form chronic infections than others and we lack a comprehensive view of the suite of genes under positive selection in the CF lung. We analyzed whole-genome sequence data from 1,000 P. aeruginosa strains with diverse ecological provenances including the CF lung. CF isolates were distributed across the phylogeny, indicating little genetic predisposition for any one clade to cause chronic infection. Isolates from the CF niche experienced stronger positive selection on core genes than those derived from environmental or acute infection sources, consistent with recent adaptation to the lung environment. Genes with the greatest differential positive selection in the CF niche include those involved in core cellular processes such as metabolism, energy production, and stress response as well as those linked to patho-adaptive processes such as antibiotic resistance, cell wall and membrane modification, quorum sensing, biofilms, mucoidy, motility, and iron homeostasis. Many genes under CF-specific differential positive selection had regulatory functions, consistent with the idea that regulatory mutations play an important role in rapid adaptation to novel environments.
Introduction
Pseudomonas aeruginosa is a ubiquitous gamma-proteobacterium commonly recovered from a wide range of niches including environmental sources such as soil and water, and various plant and animal hosts. In humans, P. aeruginosa is an opportunistic pathogen causing acute infections and chronic respiratory tract infections in patients with the genetic disorder cystic fibrosis (CF). Approximately 65–70% of adult CF patients carry P. aeruginosa and chronic infection is the leading prognostic indicator of morbidity and mortality in this patient population (Aaron et al. 2010; LiPuma 2010). Most chronic infections are acquired through colonization from environmental sources, although highly transmissible epidemic strains are an important source of patient-to-patient transmission which, in combination with the spread of multidrug resistance (Miyoshi-Akiyama et al. 2017; Parkins et al. 2018), contributes to growing public health concerns around this pathogen for both CF and non-CF patients alike.
The factors responsible for the transition from environmental strain to chronic endobronchial infection in CF patients remain poorly understood. Pseudomonas aeruginosa is believed to exist as a large, global, recombining population where individual strains opportunistically colonize the CF airways. There seems to be little association between a strain’s genotype and the niche of its isolation source (Pirnay et al. 2009; Kidd et al. 2012), with the exception of epidemic strains, suggesting that the ability to infect humans is not a characteristic of specific P. aeruginosa lineages. Permanent establishment in the CF airway is thought to be the result of rapid, mutation-driven adaptation within the host without substantial genetic exchange between strains from different hosts or environmental sources (Hauser et al. 2011; Folkesson et al. 2012). Hallmark phenotypic changes associated with chronic infection include loss of motility, reduced expression of virulence factors, mucoidy, and tolerance to stresses including antibiotics and immune system attack. These putatively patho-adaptive trait changes are underlain by a suite of genetic changes resulting from within-host evolution over the course of an infection (Smith et al. 2006; Yang et al. 2011; Diaz-Caballero et al. 2015; Marvig et al. 2015; Winstanley et al. 2016; Bartell et al. 2019). However, whole-genome population-level analyses to date have been restricted mainly to clinical isolates, so little is known about which of these genetic or functional pathways are uniquely associated with P. aeruginosa adaptation to the CF respiratory tract.
Here, we leverage the wealth of publicly available genome sequence data for P. aeruginosa derived from different niches to study niche-specific adaptation during the development of chronic infections in CF patients. Significant genomic resources for P. aeruginosa have accumulated over the last decade, with international sequencing consortia and online databases now providing public access to over 1,000 genomes from both clinical strains isolated from human infections and environmental sources (Freschi et al. 2015, 2019; Kos et al. 2015; Shrestha et al. 2017). We focus attention on two outstanding issues regarding the origin and development of chronic infections in CF patients. First, we evaluate the relationship between phylogenetic structure and ecological niche, to test whether CF isolates are derived from independent lineages across the phylogenetic spectrum of the species. Second, we compare the strength and distribution of selection pressures across the genome for three different niches (CF, acute, and environmental) to identify genes with greatest evidence for differential positive selection in the CF niche, without any a priori knowledge of their potential function or importance. We then survey the functions of these CF-selected genes, perform enrichment analyses to identify overrepresented functional classes or pathways, and discuss their potential relevance to CF-P. aeruginosa pathosystem.
Results and Discussion
A 1,000-Strain Phylogeny of P. aeruginosa Isolated from Diverse Sources
We constructed a phylogeny of 1,000 strains of P. aeruginosa isolated from diverse sources including the environment (En), acute infection of a nonhuman animal (An), acute infection of a human (Ac), and chronic infection of CF airways (CF). First, we identified 1,523 genes present in all 1,000 analyzed P. aeruginosa genomes, deemed “global strict-core” genes (supplementary table S1, fig. S1, and supplementary text, Supplementary Material online). To reduce the computational requirements for maximum likelihood (ML) tree construction, every fourth global strict-core gene was chosen to create a subset of 381 genes for alignment (357,509 nucleotides long, 52,355 single-nucleotide polymorphisms) and phylogeny building (supplementary table S2, Supplementary Material online). Phylogenetic trees constructed using ML and Bayesian methods (fig. 1A) revealed five main phylogenetic groups, named Phylogroups 1–5 following Freschi et al. (2019). The majority of strains fall within Phylogroup 1 (741 strains), which includes the common laboratory strain PAO1 and multiple epidemic CF strains, or Phylogroup 2 (238 strains), which includes the well-studied laboratory strain PA14 and is more phylogenetically diverse than the larger Phylogroup 1 (fig. 1C;supplementary table S3, Supplementary Material online). The remaining 21 strains fell into one of three less-sampled basal groups, the most divergent being Phylogroup 3 that harbors the taxonomic outlier strain PA7. Although the three basal phylogroups were significantly supported, the exclusive monophyly of the larger Phylogroups 1 and 2 received mixed support from ML and Bayesian methods, due to the inconsistent and poorly supported placement of a small number of early diverging, long-branched taxa (marked with asterisks on fig. 1A). When these early diverging lineages are excluded from tree reconstruction, the monophyly of Phylogroups 1 and 2 was significantly supported (92% ML bootstrapping and 1.0 Bayesian posterior probabilities for both; trees not shown).

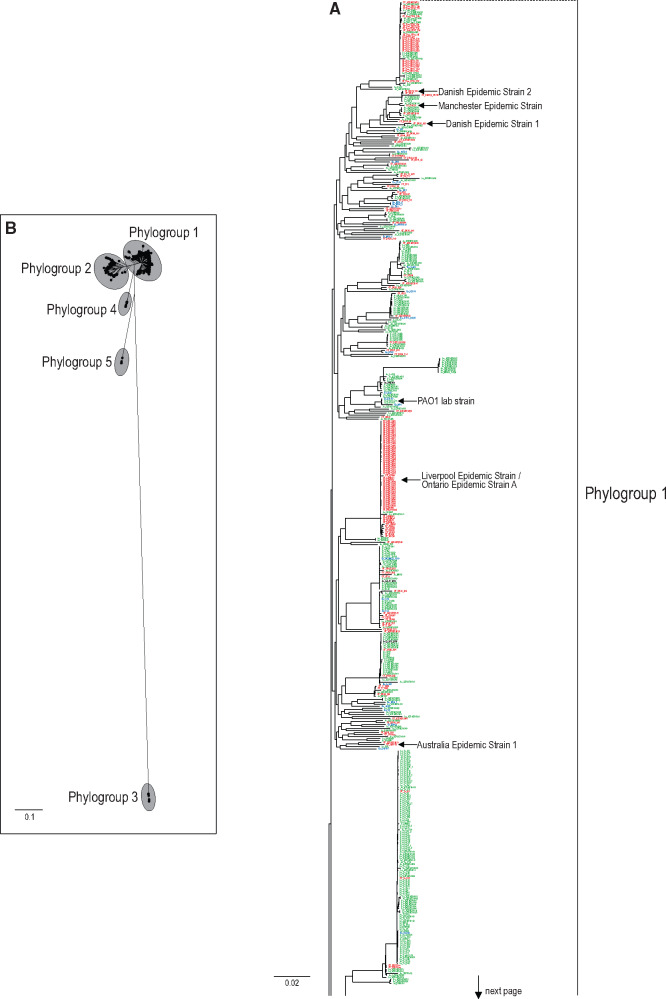
(A) ML tree constructed from alignment of 381 strict-core genes. Selected branches along the tree backbone have ML bootstrap percentages and Bayesian posterior probabilities shown above and below branches, respectively. An “X” indicates the branch was not present in the Bayesian consensus tree. Taxon names are colored to indicate source niche (red = CF, green = acute, blue = environment, black = animal). Asterisks indicate taxa that were removed to test their effect on support of main nodes. Branch leading to Phylogroup 3 is reduced to one-tenth of actual length for display purposes. (B) Unrooted ML tree with unmodified branch lengths. (C) Distribution of strain sources per phylogroup. (D) Observed strain numbers per niche compared with expected strain numbers per niche based on overall proportions in the clone-corrected strain set. Values of 1.0 indicate observed equals expected. Data are shown only for the two phylogroups with the largest sample sizes.
Ecological Niches and Phylogenetic Structure
The distribution of P. aeruginosa strains across the phylogeny reveals little association between phylogenetic structure, including phylogroups, and ecological niche (fig. 1A and C). Lineages tend not to be specifically associated with a niche, and isolates from the same niche could belong to different phylogroups or smaller subgroups/clades within phylogroups. For example, the 77 En and 17 An strains were distributed fairly evenly across phylogeny. Furthermore, the extent of within-niche diversity was similar across niches, despite the fact that CF strains likely are derived from a more similar set of habitat conditions—the CF respiratory tract—than strains from the acute niche (sampled from infections of burns, wounds, blood, eyes, ears, and urinary tracts) and the environmental niche (sampled from soil, oceans, rivers, and plant hosts). These results demonstrate that the ability to infect humans is not lineage-specific, and there are no human-specific pathovars that cause the majority of clinical infections. Moreover, our results suggest that any lineage or phylogroup can develop into a chronic infection of the CF respiratory tract, consistent with the assumption that most chronic CF infections originate from colonization by environmental strains. These results notwithstanding, there is some evidence for phylogenetic structure among CF strains, especially those causing epidemic infections. CF strains were overrepresented in Phylogroup 1 (124% of expectation) and underrepresented in Phylogroup 2 (31% of expectation), whereas the converse was true for Ac strains (chi-square value 47.3, P < 0.00001). This result is not due to biased sampling of high-frequency strains, as could be the case if the same clinical strain is sampled repeatedly from the same location (ward or hospital) at the same time. Restricting the analyses to a clone-corrected sample of 503 strains (supplementary text, Supplementary Material online) showed that that original results were robust: CF strains were significantly overrepresented (114%) and underrepresented (51%) in Phylogroups 1 and 2, respectively (fig. 1D, chi-square value = 9.095, P < 0.0026).
All epidemic CF strains fell within Phylogroup 1 (fig. 1A), including a cluster of 58 isolates corresponding to the CF-specific Liverpool Epidemic Strain/Ontario Epidemic Strain A (Aaron et al. 2010; Dettman et al. 2013). Phylogroup 1 also contained the most common epidemic CF strains from other locations, including Ontario Epidemic Strain B, Australia Epidemic Strain 1, Manchester Epidemic Strain, and Danish Epidemic strains (Parkins et al. 2018). This result suggests that Phylogroup 1 strains are more likely to develop into epidemic strains that become highly transmissible from patient to patient. Moreover, the polyphyletic origins of the epidemic clades suggest that high transmissibility is a trait, or more likely a suite of traits, that evolved independently multiple times.
Inferring the History of Selection through Estimates of Omega
The CF lung is thought to impose strong selection on P. aeruginosa because it represents a novel environment to which most colonizing strains, being derived from either the environment or acute infections, are mal-adapted. This recent history of adaptation should leave a signature of strong positive selection on some genes from CF isolates compared with those from non-CF niches. To test this prediction, we estimated the ratio of nonsynonymous to synonymous substitution (omega, or dN/dS; Yang and Bielawski 2000) for each of ∼5,510 genes constituting the “relaxed-core” gene set per niche (i.e., present in ≥95% genomes, supplementary table S4, supplementary text, Supplementary Material online), representing 92% of the average P. aeruginosa gene complement of ∼6,000 genes. Selection analyses were performed on subsets of ≤100 strains per niche (CF, n = 100; Ac, n = 100; En, n = 77; supplementary table S1, Supplementary Material online) in order to balance the breadth of phylogenetic diversity within each niche, including representation from each major clade, against the need for a computationally feasible data set.
The distribution of omega values from all 16,530 gene alignments (fig. 2A) has a mean of 0.132 (SE = 0.003), similar to that described in previous comparative genomics studies of P. aeruginosa (Dettman et al. 2013, mean = 0.10; Mosquera-Rendón et al. 2016, median = 0.1). The distribution is strongly left-skewed, with 61.4% of omega values ≤0.1, implying that the majority of genes in the P. aeruginosa genome have a history of predominantly purifying selection. Notably, mean omega was higher for the CF niche overall (ANOVA, F = 3.75, P < 0.024) and in pairwise tests against both the Ac and En niches (fig. 2B;supplementary table S5, Supplementary Material online; Tukey–Kramer HSD P < 0.047 for both), even following log10-transformation for normality (fig. 2C, ANOVA, F = 4.22, P < 0.015, Tukey–Kramer HSD P < 0.033 for both), consistent with strains from the CF niche having experienced a more recent and/or stronger history of positive selection than those from the Ac or En niche.
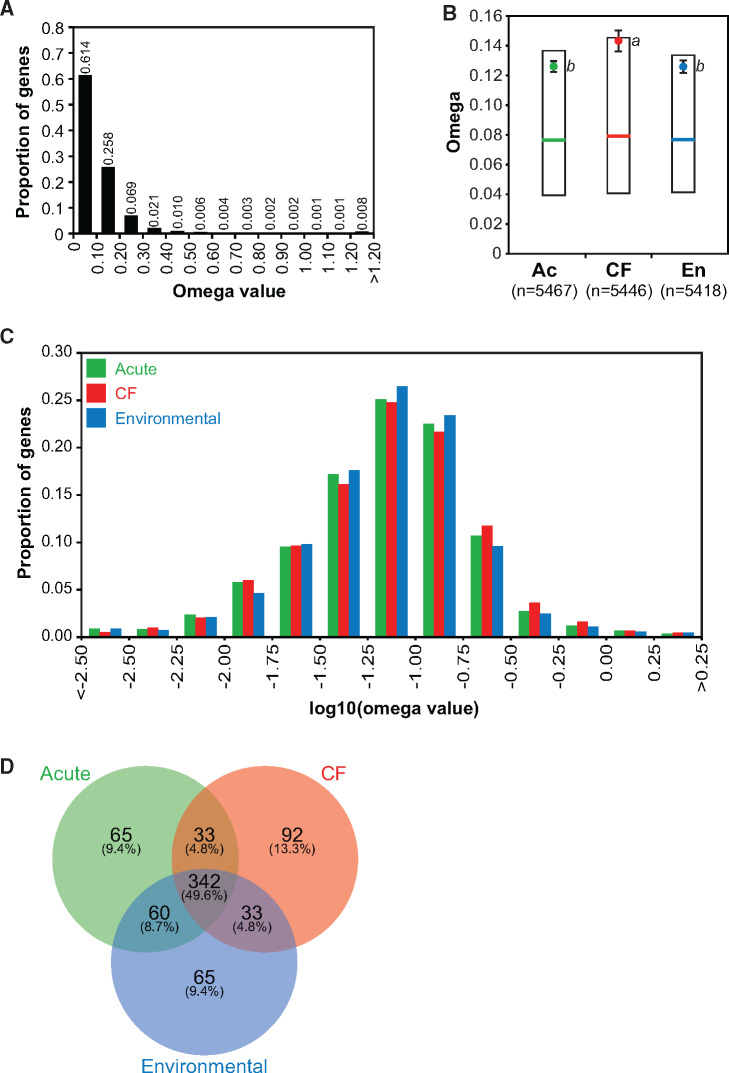
(A) Distribution of genes within binned ranges of omega values, for all data combined. Proportions are indicated above columns. (B) Comparison of omega values per niche. Circles indicate means, error bars indicate standard error, and lowercase letters indicate means that are significantly different from each other based on a Tukey–Kramer HSD test. Bars indicate medians and boxes indicate first and third quartiles. (C) Distribution of genes within binned ranges of log10-transformed omega values, presented per niche. (D) Venn diagram displaying overlap between the top-500 highest omega gene lists for each niche.
Genetic Targets of Positive Selection across Niches
Our genomic sampling scheme affords a unique opportunity to examine the spectrum of shared and unique genetic targets of positive selection across niches. The union of lists of the top 500 genes (top ∼9%) with the highest omega values in each niche resulted in a total of 690 genes, 342 (49.6%) of which were shared among all three niches (intersection, fig. 2D). Gene ontology (GO) term combined graphs revealed these shared genes participated in various biological processes, such as transport, gene expression, and biosynthesis of macromolecules (supplementary fig. S3A, Supplementary Material online), although none of the GO terms was significantly enriched after false discovery rate (FDR) correction (supplementary fig. S3D, Supplementary Material online). Genes displaying high positive selection in all three niches commonly had functions in protein secretion (type III: psc and pcr genes; type II: hxc genes) and pilus organization, in particular, fimbriae assembled by the chaperone–usher pathway (cup genes; supplementary table S6, Supplementary Material online).
When comparing top-500 omega lists, the CF niche had more unique genes experiencing relatively strong positive selection than either the Ac or En list, who together shared more genes with each other than with the CF list (fig. 2D;supplementary table S7, Supplementary Material online). GO term combined graphs of the 92 genes unique to the CF list revealed similar biological processes as the shared list (e.g., transport, gene expression) and additional processes such as RNA metabolism and oxidation–reduction (supplementary fig. S4A, Supplementary Material online). Well-represented molecular functions included oxidoreductase activity and substrate binding for nucleic acids, ATP, metal ions, and drugs (supplementary fig. S4B, Supplementary Material online); however, no GO terms were significantly enriched after FDR correction due to low samples sizes (supplementary fig. S4D, Supplementary Material online). Notably, different genes from a single operon for phenazine biosynthesis (phzA1-G1) were under strong positive selection in each niche (CF, phzA1; Ac, phzF1; En, phzE1; supplementary table S7, Supplementary Material online), demonstrating how different parts of the same pathway may be under positive selection in different niches. Phenazine production and type III secretion are considered virulence factors for P. aeruginosa (Filloux 2011; Recinos et al. 2012) and are typically studied in the context of CF infections. Our results confirm that both pathways are positively selected in the CF niche but suggest that selection on these systems is not CF specific, with these functions playing a role in adaptation more broadly.
Differential Positive Selection between Niches
Which genes are under stronger positive selection in the CF niche? To answer this question, absolute omega values were converted to relative omega values by calculating standardized Z-scores within each niche, then differential selection at each gene i (ΔSi) was calculated as the pairwise difference in Z-scores between two niches [ΔSi(niche1 − niche2) = Zi(niche1) − Zi(niche2)]. Comparing relative omega values across niches allows us to sidestep complications associated with interpreting absolute values of omega, such as when gene-wide omega is <1 despite strong positive selection on a small number of amino acid sites within the gene (Bielawski and Yang 2001; Rodrigue and Lartillot 2017), or when reduced divergence of within-species population samples like ours underestimates the strength of positive selection (Kryazhimskiy and Plotkin 2008).
We then considered three scenarios representing possible routes to the development of chronic infection in the CF niche: 1) Stepwise evolutionary progression from environmental strain to acute infection strain to chronic CF infection strain (En→Ac→CF), such that the top 500 genes with the highest ΔS(CF−Ac) values represent those with the greatest positive selection in the CF niche relative to the Ac niche; 2) Environmental strains transition directly into either acute infection strains or CF infection strains (En→Ac or En→CF, but not Ac→CF), implying that the selective pressures experienced by Ac strains are independent of those experienced by CF strains. To identify genes that had greater relative positive selection in the CF niche but not in the AC niche, we determined the top 500 genes with the greatest difference between ΔS(CF−En) and ΔS(Ac−En); 3) No evolutionary directionality, with CF strains being uniquely adapted to the CF respiratory tract independently of their niche of origin, revealed as those genes with the top 500 positive CF residuals from a multiple regression of log10-transformed CF omega values against En and Ac omega values. There is no clear consensus on which scenario is the most suitable for modeling the transition to a CF strain. For example, the phylogenetic tree indicates that CF strains can share a most recent common ancestor with Ac or En strains, suggesting that the first scenario (En→Ac→CF) and second scenario (En→CF and Ac→CF) are both applicable. To ensure that our conclusions were not heavily skewed by one scenario, we took the liberal approach of creating a union of the three lists to find the 724 genes that were identified by at least one metric (supplementary table S8, Supplementary Material online), hereafter referred to as “CF-selected” genes.
Functions of CF-Specific Differentially Positively Selected Genes
High-level combined graphs of GO term annotations (supplementary fig. S5A, Supplementary Material online) revealed that the 724 CF-selected genes were involved in biological processes such as response to stimulus, transport, gene expression, and metabolism of nucleic acids and proteins. Well-represented molecular functions included transmembrane transport, catalytic activity (oxidoreductase, hydrolase, and transferase), and binding of various substrates, such as nucleic acids, ATP, metal ions, and drugs (supplementary fig. S5B, Supplementary Material online). Statistical comparison of GO terms for the 724 CF-selected genes against all genes revealed significant enrichment for 214 GO terms (supplementary table S9A, Supplementary Material online), or 62 when reduced to the most-specific GO terms (supplementary table S9B, Supplementary Material online). Many of the significantly enriched biological processes (fig. 3) have known links to P. aeruginosa’s pathogenic lifestyle, the most obvious example being “pathogenesis,” and other processes related to cellular homeostasis, response to stress, cell motility, and biofilm formation that can affect pathogenic potential. Similar results were obtained using the more conservative approach of analyzing only the 306 CF-selected genes that were identified by all three differential selection metrics (list intersection, supplementary table S8, Supplementary Material online). Significantly enriched GO annotations largely mirrored the more liberal, union-based approach (supplementary fig. S6, Supplementary Material online), with the addition of a few notable functions, such as alginic acid biosynthesis.
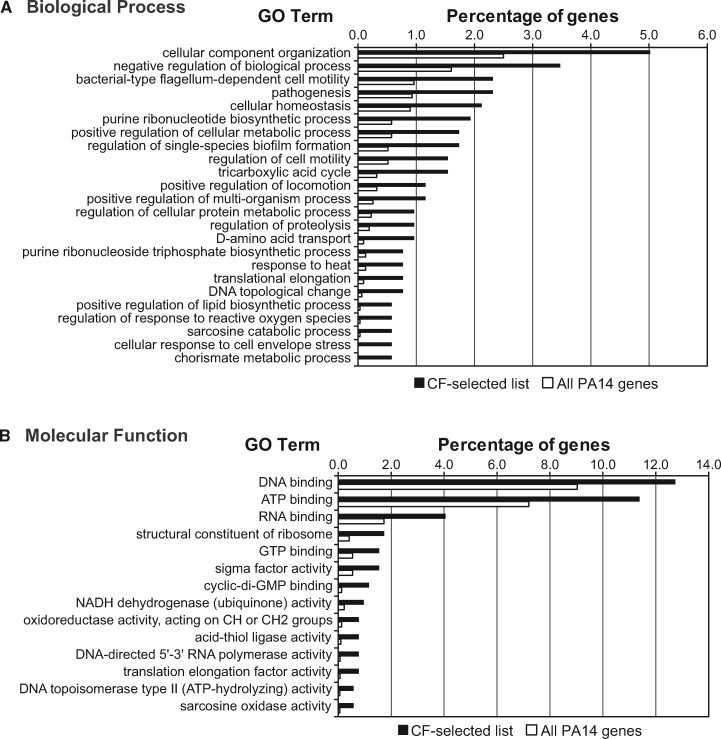
Bar graphs displaying GO terms (reduced to most specific) that were enriched in the 724 genes with high differential positive selection in the CF niche. (A) Biological processes. (B) Molecular functions. Only GO terms that were significant after FDR correction, and had three or more gene entries, are shown (see supplementary table S6, Supplementary Material online, for all GO terms and the “Cellular Component” data).
Pathways of Adaptation to the CF Lung
We supplemented the often-incomplete GO annotations with manual annotation using BLAST, homolog, and literature searches. Integrating this additional information (supplementary table S8, Supplementary Material online) with our previous analyses (fig. 3; supplementary fig. S5 and table S9, Supplementary Material online) produced a comprehensive picture of the spectrum of genetic targets under differential positive selection in the CF airway. We summarize these results in figure 4 and briefly discuss the relevance of these pathways, as well as key genes involved (supplementary table S10, Supplementary Material online), to the evolutionary adaptation of P. aeruginosa to the CF niche.
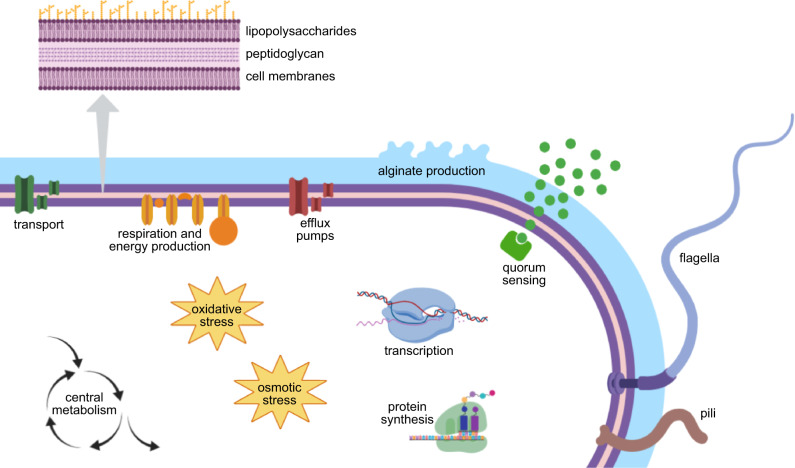
Processes and functions that were common to genes under CF-specific differential positive selection.
Central Metabolic Pathways
The complex, amino acid-rich, nutritional conditions of the CF lung are thought to provide ample opportunity for resource specialization (La Rosa et al. 2018) and adaptive diversification (Schick and Kassen 2018). Consistent with this hypothesis, key regulators of preferential carbon source utilization (Sonnleitner et al. 2012), including crc (catabolite repression control) and the CbrAB two-component system (cbrA), as well as nitrogen utilization (ntrB, ntrC), were under strong differential positive selection in the CF niche. Additional metabolic targets included genes in the glycolysis and pentose phosphate pathways (fda, eno, acsA, kguK) and those involved in transport and metabolism of branched chain amino acids (bra and bkd genes). The regulation of central metabolism has been linked to expression of P. aeruginosa virulence factors such as biofilm formation, cytotoxicity, and antibiotic resistance (Yeung et al. 2011), suggesting a high level of pleiotropy among these putatively patho-adaptive traits.
Respiration and Energy Production
The combination of thick, viscous mucus of the CF airway and growth in structured biofilms leads to greatly reduced oxygen availability to the pathogen. Evidence suggests that P. aeruginosa adapts to such oxygen limitation by not only upregulating anaerobic respiration and fermentation (Hoboth et al. 2009; Dettman et al. 2013) but also increasing the uptake of oxygen for aerobic respiration (La Rosa et al. 2018). We find evidence for adaptation via both pathways in our data set. Genes involved in anaerobic respiration through the denitrification pathway (Schobert and Jahn 2010) such as nitrate and nitric oxide reductases (nar and nor genes) were under strong differential selection. Suites of CF-selected genes associated with aerobic respiration ranged from electron donors for the generation of NADH during the tricarboxylic acid cycle (citrate, gltA; malate, mqoA; succinate, suc and sdh genes; Riquelme et al. 2019), seven subunits of the NADH dehydrogenase complex (nuo genes), components of the electron transport chain (wrbA, ubiE), cytochromes (cox, cco, cyo, and ccm genes), to those for ATP synthesis (atp genes). Our results suggest that, as P. aeruginosa adapts to the energetic conditions of the CF lung, modification of aerobic respiration is a more common adaptive solution than the anaerobic pathway.
Cellular Homeostasis and Stress Response
Multiple sources of stress in the CF airway can impose strong selection for the maintenance of cellular homeostasis in P. aeruginosa. Genes from multiple stress response pathways, including DNA repair (recA, uvrB), stringent response (relA, spoT, obgE), heat shock (htpX), and universal stress proteins (uspA), were among those under strong CF-specific differential positive selection. In particular, stable maintenance of redox balance is required to remediate multiple oxidative stressors, such as respiration-derived reactive oxygen species production, host macrophage-derived oxidative burst, and antibiotics with redox-related mechanisms of action. Consistent with previous work (Dettman et al. 2013), our CF-selected gene list contained many genes associated with maintaining redox homeostasis in response to oxidative stress (msrB, nrdA, nrdB), including the central oxidative stress regulator, oxyR, and several genes it regulates (ahpC, ankB, dps, rpsL; Wei et al. 2012). Other oxidative stress-associated genes included multifunction redox proteins such as thioredoxins, ferredoxins, glutaredoxins, and glutathione S-transferases, respiration-related genes such as cytochromes (see previous section), and over 20 additional genes with clear redox functions such as oxidases, reductases, dehydrogenases, and dioxygenases. The accumulation of viscous mucus, along with electrolyte imbalances due to defective ion transport in the host, leads to increased osmotic stress in CF airway secretions (Henderson et al. 2014). We find several genes involved with osmoregulation under differential positive selection (osmC, opgG, aqpZ), including the sensor and response regulator (envZ and ompR; Cai and Inouye 2002) of the central regulator of osmotic stress response (also known as amgS/amgR). Notably, CF-specific selection was found on the pathway that produces the potent osmoprotectant glycine betaine (Wargo 2013), including six genes acting on three different substrates in this pathway (choline [betA, betI], glycine betaine [gbcB], and sarcosine [soxB, soxD, soxG]), and two subunits of the glycine betaine transporter itself (cbcV, cbcW). Maintaining cellular homeostasis in the face of multiple sources of stress thus appears to be an important adaptive strategy for the survival of P. aeruginosa in CF lungs.
Antibiotic Resistance
Treatment regimes for CF patients typically include regular doses of antibiotics to control infection, so it is no surprise that our analyses identified multiple CF-selected genes associated with antibiotic resistance. Relevant targets include genes implicated in resistance to quinolones (gyrA, gyrB, topA, parE; Bruchmann et al. 2013), beta-lactams (ftsI, penicillin-binding protein 3; Jorth et al. 2017), aminoglycosides (amiB, hflC; Hinz et al. 2011), and general resistance through multidrug efflux, such as mexA from the MexAB-OprM efflux pump and mexT, the regulator of the MexEF-OprN efflux pump (Köhler et al. 1999). We also find many genes associated with transcription and protein synthesis, which are inhibition targets for tetracyclines, aminoglycosides, and macrolides (Arenz and Wilson 2016; Ma et al. 2016). Examples include three subunits of RNA polymerase core enzyme (rpo genes), transcriptional elongation and anti/termination factors (rho, gre genes, and nus genes), translation elongation factors (fusA1, tsf, tufA, tufB), and multiple proteins that comprise the 30S and 50S ribosomal subunits (rps and rpl genes, respectively). Note that most genes conferring antibiotic resistance play important roles in other essential cellular processes, so the source of selective pressure on the aforementioned genes may be a composite of antibiotic resistance and other factors experienced in the CF lung.
Cell Wall and Membranes
Many genes related to the cell wall and outer membrane showed evidence of CF-specific positive selection, although it is not always clear whether the selective cause is the integrity of these structures themselves, or resistance to the manifold challenges of the CF lung such as high osmotic stress. CF-selected genes included several involved in the synthesis (acc genes) and metabolism (fad and fab genes; Fujita et al. 2007) of fatty acids, a component of membrane phospholipids. We find a number of genes associated with lipopolysaccharides (wapH, lpxO2, galU), a main constituent of the outer membrane of Gram-negative bacteria that is important as a virulence factor (endotoxin) and in recognition by the host’s adaptive immune response (Hauser et al. 2011). The biosynthesis of peptidoglycan, a main component of bacterial cell walls, is inhibited by beta-lactam antibiotics and selection for resistance often manifests as modification in the targeted pathway, as evidenced in our sample by CF-selected genes, such as glmM, oprL, erfK, murC, and uppS.
Alginate and the Mucoid Phenotype
Conversion to mucoid phenotype is a common adaptive change observed in P. aeruginosa strains that chronically infect CF lungs. The overproduction of alginate creates an extracellular matrix protecting bacteria from the host immune response, antibiotic activity, and environmental cell envelope stressors (Hauser et al. 2011). Our CF-selected list included several components of the alginate pathway (alg genes), as well as its main regulators (mucA, algR, algU). The algU gene (also known as algT) encodes a sigma factor that binds with RNA polymerase to specifically activate alginate biosynthesis genes, whereas mucA encodes the anti-sigma factor that sequesters algU and prevents alginate production. Mutations in mucA and algU are the most common causes of mucoidy and reversion to nonmucoidy, respectively (Caçador et al. 2018).
Quorum Sensing, Biofilms, and Motility
Cell-to-cell communication via quorum sensing allows bacteria to synchronize behaviors based on population density. This process impacts diverse phenotypes linked to pathogenicity and virulence, such as stress responses, mucoidy (see above), biofilm formation, and motility (Miller and Bassler 2001), which are coordinated by a complex, hierarchical regulatory network (Lee and Zhang 2015). Our analyses found that the master regulators of each of the three main systems regulating quorum sensing and virulence factors in P. aeruginosa (las, rhl, and pqs systems) were under CF-specific differential positive selection: lasR, rhlR, and pqsR (mvfR) genes, respectively. In addition, many transcriptional regulators of quorum sensing, biofilm formation, and motility phenotypes were also CF-selected (dksA, gacA, rsmN, vfr; sigma factors rpoN and rpoS). Other genes with presumably more phenotype-specific effects were those for synthesis of quorum-sensing signaling molecules (lasI, pheA, phnB) and secondary regulation (cysB, pmpR), metabolism of the signaling molecule cyclic diguanylate important for biofilm formation and mucoidy (siaD, bifA, rbdA, cmpX), and the synthesis of flagella (flg, fli, and fle genes) and pili (pil genes). In addition to roles in motility and adhesion, flagella and pili are also immunogenic structures, so selection for diversity may assist P. aeruginosa in evading the adaptive immune response.
Iron Homeostasis
Iron is often scarce in the lung because it is chelated or bound to hemoproteins, so mechanisms for acquiring sufficient iron, such as iron-scavenging siderophores, are considered virulence factors for most bacterial pathogens (Minandri et al. 2016). Although only two siderophore-related genes showed evidence of differential positive selection in the CF niche (pyochelin, pchB; pyoverdine, pvcB), many other genes on our list were involved in iron transport, storage, or homeostasis (ferric enterobactin, fepD; bacterioferritin, bfrB; femI). A suite of genes for bacterial heme biosynthesis and transport (phuU and hem genes) were also CF-selected, suggesting that heme synthesis may be as important as heme acquisition from host sources (Choby and Skaar 2016). Given that many redox and stress response proteins from the previous sections require iron–sulfur clusters or heme moieties as cofactors, and heme is an essential component of the respiratory cytochromes discussed above, the maintenance of intracellular iron homeostasis may be under selection by multiple sources in P. aeruginosa.
Regulation
A common theme emerging from our analyses is that differential positive selection on the regulation of various pathways or processes makes an important contribution to CF-specific adaptive evolution. Indeed, 42% (10/24) of the significantly enriched GO terms in figure 3A involved “regulation,” and the CF-selected regulatory genes were often high-level master regulators, such as nine different sigma factors that each affect the expression of potentially thousands of downstream genes. We also found at least 46 transcriptional regulators from families such as AraC, GntR, LysR, MarR, RpiR, and TetR, and at least 13 sensors or response regulators of two-component systems that regulate multiple virulence factors and antibiotic resistance in P. aeruginosa (Gooderham and Hancock 2009). These results are not unusual: Regulatory mutations are commonly recovered from the early stages of adaptation to novel, stressful laboratory environments in vitro, presumably because they restore the balance between stress responses and vegetative growth (Ferenci 2005; Dettman et al. 2012). If correct, this interpretation suggests that the early stages in the development of chronic infections can be understood as a special case of adaptation to the novel, stressful conditions of the CF lung.
Unknown Functions
The above discussion is heavily biased toward named genes that already have well-characterized, or at least putative, functions (supplementary tables S8 and S10, Supplementary Material online). Of the 724 genes on our CF-selected list, we were able to find gene names for only 405 (56%) of them. Many unnamed genes have homology to known protein families, whose functions fall clearly into some of the relevant categories above. Even more interesting may be the 224 CF-selected genes that are listed as “hypothetical proteins.” After various homolog and database searches, we could not determine even a putative function for 138 of these genes.
Conclusions/Summary
Our results provide three important insights into the P. aeruginosa/CF pathosystem and the natural history of selection during in vivo evolution. First, isolates forming chronic infections are, with the exception of the highly transmissible epidemic strains, derived from multiple sources that likely include both acute infections and the environment. Our analyses of whole-genome sequence data from a broad sampling of 1,000 diverse P. aeruginosa strains support previous inferences on the population structure of this opportunistic pathogen (Pirnay et al. 2009; Kidd et al. 2012; Dettman et al. 2013; Grosso-Becerra et al. 2014). Our increased sampling power confirms the “non-clonal epidemic” nature of P. aeruginosa, in which there exists a background of genetically diverse strains from which CF-adapted strains, including highly successful epidemic clones, occasionally arise. This result suggests that isolates from chronic CF infections can be understood as examples of convergent evolution resulting from diverse genetic sources and evolving more or less independently from each other within a single host.
Second, strains from the CF niche have experienced a stronger or more recent history of positive selection than strains from acute or environmental niches. A growing body of literature compares isolates sampled longitudinally through time from individual CF patients to document the changes occurring over the course of an infection (Smith et al. 2006; Yang et al. 2011; Diaz-Caballero et al. 2015; Marvig et al. 2015; La Rosa et al. 2018; Bartell et al. 2019). Although these studies provide insight into the spectrum of genetic and regulatory changes occurring during chronic infection, they cannot by themselves make strong inferences regarding the strength of selection. Our results support the assumption that the CF lung represents a novel environment for isolates of P. aeruginosa from diverse sources, and that adaptation to the conditions of the CF lung is a process that is essential for the establishment of chronic infections. Furthermore, this finding validates the main goal of many previous studies, which is to identify the patterns and mechanisms of adaptive evolution that occur during the transition from a free-living, environmental strain to one that chronically infects CF lungs (Hauser et al. 2011; Folkesson et al. 2012). Why P. aeruginosa can adapt more readily than any other microbial species colonizing the CF lung remains unresolved.
Third, our study is the first to make direct comparisons between large samples of P. aeruginosa strains collected from different ecological niches, allowing us to distinguish CF-specific selection from selection across all niches, and to challenge hypotheses from previous work that focused only on clinical strains. Studies relying on longitudinally sampled CF strains can make inferences about the genetic targets of selection; however, different strains often take distinct evolutionary paths to reach convergent patho-adaptive phenotypes, even within the lungs of a single CF patient (La Rosa et al. 2018). To overcome this problem, we performed comparative analyses of a broad, cross-sectional sample of strains that have already adapted to individual CF-patient lungs, each with complex and potentially unique conditions. Our approach relies on the signatures of selection summed over the multiple independent evolutionary paths taken by a diverse set of strains that have each successfully adapted to form chronic infections of CF airway niche, and contrasts these results against those that were isolated from distinct, non-CF niches. Our analyses identified multiple pathways under differential positive selection the CF niche: 1) central metabolism; 2) respiration and energy production; 3) cellular homeostasis and stress response; 4) antibiotic resistance; 5) cell wall and membranes; 6) alginate and mucoidy; 7) quorum sensing, biofilms, and motility; 8) iron homeostasis; and 9) regulation. Many of these pathways have been previously implicated in the adaptation of P. aeruginosa to the CF airways (Hauser et al. 2011; Folkesson et al. 2012). Importantly, a large number of CF-specific genes we objectively identified here have independent corroborating evidence for positive selection and roles in adaptation (e.g., antibiotic resistance), providing proof-of-concept for our novel, blind approach. Conversely, many genes have no known relation to the P. aeruginosa/CF pathosystem, and some have no known function at all. Our analysis has generated an objective list of prime candidates for further investigation of their roles in the CF-specific adaptation of P. aeruginosa, regardless of a priori knowledge.
Our study provides a unique view of the phylogenetic sources of P. aeruginosa isolates colonizing the CF lung and the spectrum of genetic pathways under selection as they undergo transition to chronic infection. The picture that emerges is one of repeated, convergent evolution by diverse colonizing strains as they adapt to the uniquely stressful conditions of the CF lung resulting ultimately in the development of chronic infections recalcitrant to further treatment. Any therapy that can prevent, or at least delay, this process would be of outstanding benefit to CF patients.
Materials and Methods
Strains and Genome Data
Genome sequence data for a collection of 1,000 unique P. aeruginosa strains were downloaded from public databases or obtained directly from authors in other research groups (supplementary table S1, Supplementary Material online). Based on the available information, each strain was assigned to a niche of origin: cystic fibrosis airways (CF; n = 229), acute infection of a human host (Ac; n = 677), acute infection of a nonhuman animal (An; n = 17), and the environment (En; n = 77).
Orthologs
A list of orthologs was compiled from OrtholugeDB (Whiteside et al. 2013) using pairwise comparisons between annotated genomes of acute strains PA14 and PAO1, CF strains LESB58 and DK2, and environmental strain M18 (supplementary text, Supplementary Material online). Presence of each of the 6,874 orthologs in all genomes was assessed using BlastN searches with a minimum expectation value (e) of 0.0001, and retaining only those with a sequence identity ≥80%, a length of ≥75 nucleotides, and a length ≥25% of the number of nucleotides in the query gene sequence.
Phylogeny Construction
All phylogenies were built from a set of 381 genes that comprised every fourth “global strict-core” gene in order along the PA14 reference genome (supplementary table S2, Supplementary Material online). Sequences were aligned using MUSCLE (version 3.8; Edgar 2004), and phylogenetic trees were built using RAxML (version 8.2.4; Stamatakis 2014) and ExaBayes (version 1.5; Aberer et al. 2014) (see supplementary text, Supplementary Material online, for details).
Selection Analyses
The 1,523 “global strict-core” genes represented only ∼25% of the average P. aeruginosa gene complement of ∼6,000 genes. To extend our screen to a larger proportion of the genome, and to include true core genes that may be missing due to incomplete draft assemblies, selection analysis was performed on “relaxed-core” genes, as defined by a ≥95% presence threshold (∼5,510 genes per niche; supplementary text, Supplementary Material online). In addition to restricting selection analysis to strain subsets from the CF, Ac, and En niches (n ≤ 100; supplementary table S1, Supplementary Material online), the An niche was excluded due to small sample size (n = 17). Selection analyses were performed using an ML approach implemented by PAML (version 4.7; Yang 2007). For each alignment, gene-wide omega (dN/dS) was calculated using the niche-specific ML tree (runmode = 0), ten rate categories (ncatG = 10), and all sites constrained to the same rate category (NSsites = 0). Recognizing that omega estimates are sensitive to even minor alignment errors, two additional quality control steps were applied. First, prior to selection analyses, all alignments were screened for proper start codons and appropriate reading frames (alignment lengths a multiple of three). Flagged alignments were manually inspected, and if necessary, edited to restore proper reading frame. Second, after selection analyses, all alignments with resulting omega values ≥0.30 were manually inspected and verified to ensure alignment accuracy.
Differential Selection between Niches
To account for differences in mean omega values between niches, standardized Z-scores were calculated within each niche as Zi = [(χi − χ)/σ], where χi is the raw omega value, χ is the sample mean, and σ is the sample standard deviation. Differential selection (ΔSi) between niches for gene i was estimated simply as the difference between Z-scores from the two niches (ΔSi(niche1 − niche2) = Zi(niche1) − Zi(niche2)). For example, highly positive Z-scores in niche 1 and highly negative Z-scores in niche 2 would indicate high differential, positive selection in niche 1. Exploratory analyses using a similar metric, the ratio of raw omega values from two niches, gave similar results and so are not reported here. For multiple regression analyses, omega values were first log10-transformed then regressed using a generalized linear model in the R statistical package (R Core Team 2013).
Gene Functions
Blast2GO (version 5.2.5; Götz et al. 2008) was used for functional analyses of GO information, based on the GO information for strain PA14, so PA14 gene names or identifiers are used here for reference. Combined graphs (direct acyclic graphs) were built to visualize the relationships and dependencies among annotated genes. Fisher exact tests were used to test whether specific GO terms were significantly enriched in the queried gene lists, given GO terms were represented by two or more genes in the query list. Correction for multiple comparisons was applied using the Benjamini–Hochberg method for calculating the FDR (Q = 0.10 for 10% false positives). When reported here, Fisher exact test P-values were significant after FDR correction unless otherwise noted by “ns after FDR.” Owing to the vertical dependence of hierarchically related GO terms, significantly enriched lists were, in some cases, reduced to the most specific GO terms.
Acknowledgments
This work was supported, in part, by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2019-05622) and Cystic Fibrosis Canada (488978).
Author Contributions
J.R.D. and R.K. designed research; J.R.D. performed research; J.R.D. analyzed data; and J.R.D and R.K. wrote the article.
Data Availability
The data underlying this article were derived from sources in the public domain or from a third party. Details of data sources for each of 1,000 genomes are provided in supplementary table S1, Supplementary Material online.