 The Genomic Selfing Syndrome Accompanies the Evolutionary Breakdown of Heterostyly
The Genomic Selfing Syndrome Accompanies the Evolutionary Breakdown of Heterostyly
Molecular Biology and Evolution



- Altmetric
The evolutionary transition from outcrossing to selfing can have important genomic consequences. Decreased effective population size and the reduced efficacy of selection are predicted to play an important role in the molecular evolution of the genomes of selfing species. We investigated evidence for molecular signatures of the genomic selfing syndrome using 66 species of Primula including distylous (outcrossing) and derived homostylous (selfing) taxa. We complemented our comparative analysis with a microevolutionary study of P. chungensis, which is polymorphic for mating system and consists of both distylous and homostylous populations. We generated chloroplast and nuclear genomic data sets for distylous, homostylous, and distylous–homostylous species and identified patterns of nonsynonymous to synonymous divergence (dN/dS) and polymorphism (πN/πS) in species or lineages with contrasting mating systems. Our analysis of coding sequence divergence and polymorphism detected strongly reduced genetic diversity and heterozygosity, decreased efficacy of purifying selection, purging of large-effect deleterious mutations, and lower rates of adaptive evolution in samples from homostylous compared with distylous populations, consistent with theoretical expectations of the genomic selfing syndrome. Our results demonstrate that self-fertilization is a major driver of molecular evolutionary processes with genomic signatures of selfing evident in both old and relatively young homostylous populations.
Introduction
The evolution of predominant self-fertilization (selfing) from obligate cross-fertilization (outcrossing) is recognized as the most frequent reproductive transition in flowering plants (Stebbins 1957). Although only ∼10–15% of angiosperm species are highly autogamous, there is extensive comparative evidence of multiple independent origins of selfing from outcrossing, particularly in herbaceous taxa (Stebbins 1974; Jain 1976; Barrett et al. 1996; Igic et al. 2006). The frequency in which predominant selfing originates in hermaphrodite organisms (Jarne and Charlesworth 1993) provides opportunities to investigate the evolution of convergent morphological (Sicard and Lenhard 2011) and genomic (Cutter 2019) syndromes. Efforts to understand how and why selfing evolves has attracted considerable interest since the early work of Charles Darwin, George Henslow, and Hermann Muller (reviewed in Lloyd 1980), and continues today employing diverse comparative, experimental, and genetic approaches (Busch and Delph 2012; Igic and Busch 2013; Wright et al. 2013; Barrett et al. 2014). Numerous theoretical models have explored the selective factors influencing variation in plant mating systems (reviewed in Goodwillie et al. 2005), and the recent development of molecular tools enables investigation of the consequences of mating-system transitions on genomic variation and molecular evolution.
Compared with obligate outcrossing, predominant selfing is expected to reduce the effective population size, Ne, because of nonindependent gamete sampling and the prevalence of genome-wide homozygosity (Pollak 1987; Nordborg 2000). When all else is equal, Ne=N/(1 + FIS), where N is the effective population size under panmixia and FIS is Wright’s inbreeding coefficient, Ne should be diminished up to 2-fold in the case of complete self-fertilization with FIS=1. Moreover, Ne is expected to be reduced further by the action of linked selection (Charlesworth et al. 1993; Kamran-Disfani and Agrawal 2014; Roze 2016), including background selection and selection sweeps, because of the restricted levels of effective recombination and higher linkage disequilibrium (LD) in selfers than outcrossers (Flint-Garcia et al. 2003). In addition, the colonizing ability of many selfers, owing to their ability to benefit from reproductive assurance, may further contribute to decreasing Ne as a result of recurrent colonization–extinction cycles and severe bottlenecks (Baker 1955; Lloyd 1980; Charlesworth and Wright 2001; Pannell and Fields 2014).
The rate of molecular evolution of mutations subject to selection is a function of Nes, the product of the effective population size and the selection coefficient (s) (Kimura 1983). As proposed by the nearly neutral theory, there should be a large fraction of slightly deleterious mutations with selection coefficients in the order of 1/Ne, thus differences in the effective population size can strongly affect the outcome of purifying selection against these mutations (Ohta 1992; Akashi et al. 2012). The reduced Ne in selfers should increase the role of drift relative to selection resulting in a higher proportion of mutations that behave almost neutrally. This process can thus lead to the accumulation of deleterious mutations and stochastic loss of beneficial mutations (“Muller’s ratchet”; Heller and Smith 1978). Assuming similar mutation rates (μ), both the ratio of nonsynonymous to synonymous polymorphism (πN/πS) (equivalently, the ratio of 0- and 4-fold degenerate positions in protein-coding sequences, π0/π4) and the degree of divergence (dN/dS) are expected to be higher in selfing than outcrossing lineages, as a consequence of the accumulation and fixation of weakly deleterious mutations. However, strongly deleterious and highly recessive mutations are more likely to be purged by selection because the homozygous genetic background of selfers limits opportunities for the masking of deleterious mutations (Wang et al. 1999; Arunkumar et al. 2015). Despite the theoretical expectation of genomic degradation under recurrent selfing, empirical evidence for selfing as an “evolutionary dead end” is mixed (Takebayashi and Morrell 2001; Igic and Busch 2013). Therefore, further comparisons of the genetics of related outcrossing and selfing plant populations to determine whether “genomic selfing syndromes” (Cutter 2019) are evident should provide further insight into why some selfing lineages persist whereas others go extinct.
Why the efficacy of purifying selection varies among species is a key question in evolutionary genetics. A recent survey of plant and animal species investigated the relation between effective population size and the efficacy of selection and found that among plants, mating system and longevity were significant correlates (Chen et al. 2017). For genes under strong purifying selection, different patterns of selection are expected between selfers and outcrossers, as indicated by a higher ratio of nonsynonymous to synonymous polymorphisms (πN/πS) and/or substitutions (dN/dS). However, these predictions are not always supported by empirical data from comparisons of species pairs with contrasting mating systems. Polymorphism-based measures have mostly supported the prediction of a reduced selection efficacy in selfers (Cutter et al. 2008; Ness et al. 2012; Arunkumar et al. 2015; Burgarella et al. 2015), and this pattern was further confirmed by comparative analysis of genome-wide polymorphism data from both plants and animals (Chen et al. 2017). However, for comparisons involving dN/dS the evidence for relaxed selection is mixed with some studies supporting weak purifying selection in selfing plants (Slotte et al. 2010, 2013; Qiu et al. 2011; Hazzouri et al. 2012) whereas others have failed to detect the reduced efficacy of selection (e.g., Arabidopsis, Wright et al. 2002; Triticeae, Haudry et al. 2008; Escobar et al. 2010; Glémin and Muyle 2014) and also in animals (e.g., Caenorhabditis, Cutter et al. 2008).
The recent origin of selfing is often invoked to explain discrepancies between theoretical predictions and empirical data (Wright et al. 2008; Glémin and Galtier 2012; Ness et al. 2012; Cutter 2019). Divergence patterns revealed in relatively young selfers may largely reflect the influence of a long history of outcrossing in the immediate ancestor. This effect is likely to be magnified when the evolution of selfing is associated with speciation, with a change in mating system evolving gradually after reproductive isolation, as may have occurred in the highly self-fertilizing Arabidopsis thaliana (Tang et al. 2007). Phylogenetically informed comparative analyses of the efficacy of purifying selection involving related selfers of contrasting age should provide opportunities to investigate changes in the genomic selfing syndrome during the evolutionary history of taxa.
The evolutionary breakdown of the sexual polymorphism heterostyly to homostyly (fig. 1a) provides rich opportunities for investigating transitions from outcrossing to selfing and their genomic consequences (reviewed in Barrett 2019). Populations with this mating polymorphism are usually composed of two (distyly) or three (tristyly) floral morphs that differ reciprocally in stigma and anther height and possess a suite of ancillary polymorphisms involving stigmas and pollen (Darwin 1877; Ganders 1979; Barrett 1992). Heterostylous populations commonly exhibit a heteromorphic self-incompatibility system that limits or entirely prevents self- and intramorph mating, thus enforcing phenotypic disassortative mating. Heterostyly is reliably reported from 28 angiosperm families and in the majority, the polymorphism has broken down giving rise to a monomorphic homostylous condition, with populations usually composed of a single, fully self-compatible floral form with anthers and stigmas close together within a flower (Ganders 1979; Weller 1992; Barrett 2019). Homostylous plants most frequently have the capacity for autonomous self-pollination and, as a result, possess high selfing rates (Piper et al. 1984; Ganders et al. 1985). In distylous taxa, homostyles can either have long styles and long-level stamens (long homostyles) or short styles and short-level stamens (short homostyles), with the former more commonly encountered in natural populations, either as intraspecific variants in distylous populations (Crosby 1949; Charlesworth and Charlesworth 1979; Yuan, Barrett, et al. 2017; Zhou et al. 2017; Shao et al. 2019), or as derived monomorphic varieties or species in otherwise distylous lineages (Schoen et al. 1997; Truyens et al. 2005; de Vos, Wüest, et al. 2014). In contrast to the relatively gradual evolution of high selfing rates via mixed mating, characteristic of most mating-system transitions in angiosperms, predominant selfing arises very rapidly in distylous taxa, by genetic changes at the S-locus linkage group governing the heterostylous syndrome (reviewed in Kappel et al. 2017).

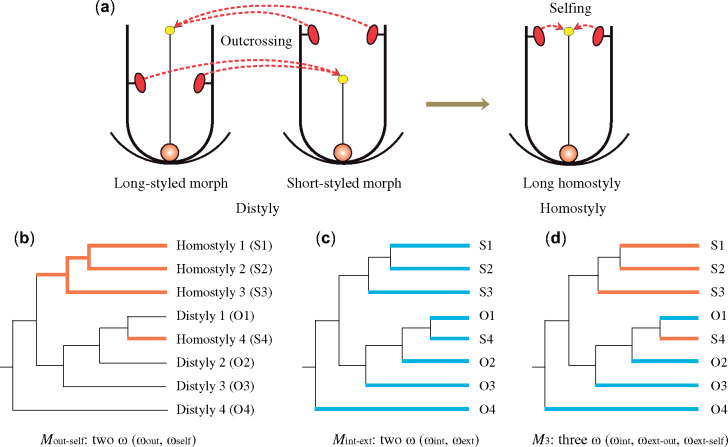
(a) Floral phenotypes associated with the breakdown of distyly to long homostyly and the mating-system transition from outcrossing to selfing that is involved. Solid arrow indicates the evolutionary transition; dashed arrows indicate predominant matings. Branch models tested with codeml in PAML analyses: (b) Mout-self: two ω for outcrossing (thin black) and selfing (thick orange) branches; (c) Mint-ext: two ω for internal (thin black) and external (thick blue) branches; (d) M3: three ω for internal branches (thin black), outcrossing (thick blue), and selfing (thick orange) external branches. Outcrossing and selfing branches on models are indicated by O and S.
Primula (Primulaceae) provides a valuable comparative system for investigating the evolutionary and genomic consequences of mating-system transitions. Beginning with Darwin’s classic work on heterostyly in Primula (Darwin 1877), the genus has received over a century of sustained interest and today is the most well-studied heterostylous taxon (Mast and Conti 2006; Gilmartin 2015). The vast majority (92%) of ∼400–500 Primula species are distylous, with the remaining species monomorphic for stylar condition and homostylous. Homostyles are distributed among 19 of the 38 sections in the genus and in all but one they co-occur with distylous species (Mast et al. 2006; de Vos, Wüest, et al. 2014). Phylogenetic analyses and ancestral state reconstructions of Primula species demonstrate a single origin of distyly but numerous independent breakdown events to homostyly (Mast et al. 2006; de Vos, Wüest, et al. 2014; Zhong et al. 2019). Significantly, intraspecific investigations of several Primula species have provided additional microevolutionary evidence that homostyles are derived from distylous morphs (Crosby 1949; Charlesworth and Charlesworth 1979; Yuan, Barrett, et al. 2017; Zhou et al. 2017; Shao et al. 2019). Thus, in Primula, the transition from outcrossing to selfing has occurred repeatedly in different lineages over a range of contrasting timescales, which should allow an evaluation of the time-dependent effects of selfing among multiple independent transitions.
Here, we employ phylogenetic and comparative genomic approaches of both nuclear and chloroplast protein-coding genes to investigate the genetic and genomic consequences of mating-system transitions in a sample of 66 species of distylous and homostylous Primula. We also conducted a detailed microevolutionary investigation of P. chungensis, because we previously demonstrated that this species is comprised both distylous, homostylous, and mixed populations containing both floral forms (Zhou et al. 2017). Contrary to most other homostylous species of Primula, the homostylous lineages of P. chungensis do not exhibit well-developed morphological selfing syndromes and probably represent more recent transitions to selfing from outcrossing than occur in fully homostylous species (Zhou et al. 2017). Thus, our sampling of homostyles provides a novel opportunity to evaluate the genomic consequences of different time scales of the history of selfing in a system in which the initial transition from outcrossing to selfing is rapid.
Our investigation specifically addressed the following questions: 1) Do homostylous species of Primula exhibit higher dN/dS ratios than distylous outcrossing species? And if so, is this pattern of divergence consistent for both chloroplast and nuclear genomes given their contrasting patterns of inheritance? 2) At the species level, does the more recently derived selfing lineage of P. chungensis exhibit higher πN/πS ratios than the ancestral outcrossing lineage? 3) Is the elevated proportion of nonsynonymous mutations observed in P. chungensis (see Results) associated with a reduced efficacy of purifying selection on nearly neutral mutations? And, in addition to relaxed selection, is there evidence for the purging effect of selfing on large deleterious mutations?
Results
Molecular Evolution of the Chloroplast Genome
The reconstructed phylogeny of 60 Primula species using the whole chloroplast genome and ML analyses is presented in supplementary figure S1, Supplementary Material online. Detailed statistics for the complete plastid genomes are provided in supplementary table S1, Supplementary Material online. Seventy-two chloroplast coding genes were evident in all 78 accessions.
For protein-coding sequences, we used the codeml implementation in PAML to apply different branch models both on individual gene alignments and the concatenated data sets (fig. 1). In the concatenated data set, the ω value was 0.220 under the null one-ratio model (hereafter M0; supplementary table S2a, Supplementary Material online), indicating that most Primula chloroplast genes evolve under purifying selection. Mout-self provided a significantly better fit than M0 (χ2=44.384, df=1, P < 0.001; supplementary table S2a, Supplementary Material online). Increased values of ω in the plastid coding region of homostylous relative to distylous branches (0.278 and 0.209, respectively) indicated that selfers experience an accelerated accumulation of slightly deleterious mutations compared with their outcrossing counterparts. Concatenated alignment data sets exhibited a statistically significant better fit to Mint-ext compared with M0 (χ2=95.200, df=1, P < 0.001; supplementary table S2a, Supplementary Material online). Higher ω values in external relative to internal branches (0.251 and 0.177, respectively) were consistent with the hypothesis that most mildly deleterious mutations are removed by purifying selection in the long term, whereas slightly deleterious mutations showed relatively more persistence over the short term. M3 provided a significantly better fit than Mint-ext (χ2=9.398, df=1, P < 0.01; supplementary table S2a, Supplementary Material online). In M3, internal branches had lower ω values (0.177) relative to the two external branches (0.240 and 0.278 for outcrossing and selfing branches, respectively) concordant with the results for Mint-ext and Mout-self.
When these genes were analyzed separately, we identified 11 (15.3%) as “evolving differentially” and 10 of 11 (90.9%) had higher ω values for selfing branches (fig. 2a). Furthermore, the results from the branch-site model identified that in selfers none of the ten fast-evolving genes showed signs of positive selection (supplementary table S3, Supplementary Material online). We identified 23 genes (31.9%) with a statistically significant better fit of Mint-ext compared with M0, based on different ω values between internal and external branches, although in most cases (22 of 23) there were higher ω values for external branches (fig. 2b). Overall, ω values for selfing and external branches were significantly higher (Wilcoxon-signed rank test, P < 0.001) resulting in a positive difference in (ωself−ωout) and (ωext−ωint) (fig. 3a).
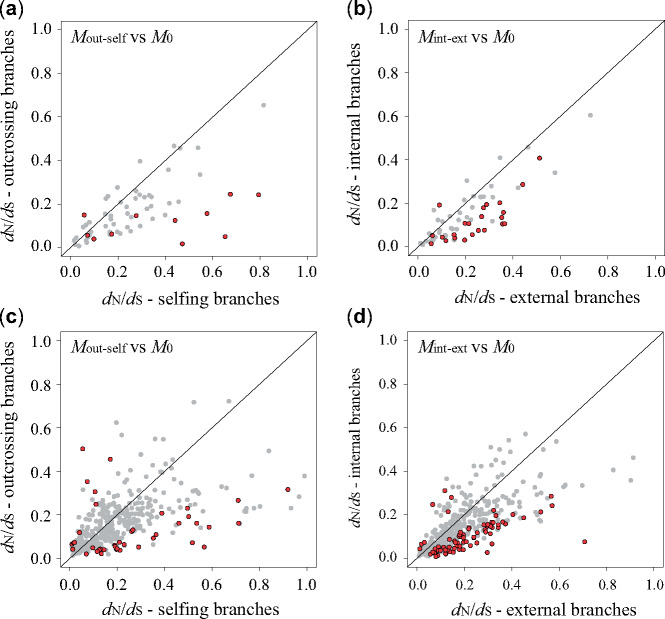
Protein evolution of 72 chloroplast genes among 60 Primula species and 445 nuclear orthologs among 12 Primula species. Plot of dN/dS values in selfing (x-axis) and outcrossing (y-axis) branches from the model Mout-self for (a) chloroplast genes and (c) nuclear orthologs, respectively. Plot of dN/dS values in external (x-axis) and internal (y-axis) branches from the model Mint-ext for (b) chloroplast genes and (d) nuclear orthologs, respectively. Genes with statistically significant differences between model Mout-self/Mint-ext and null model (M0) are in red.
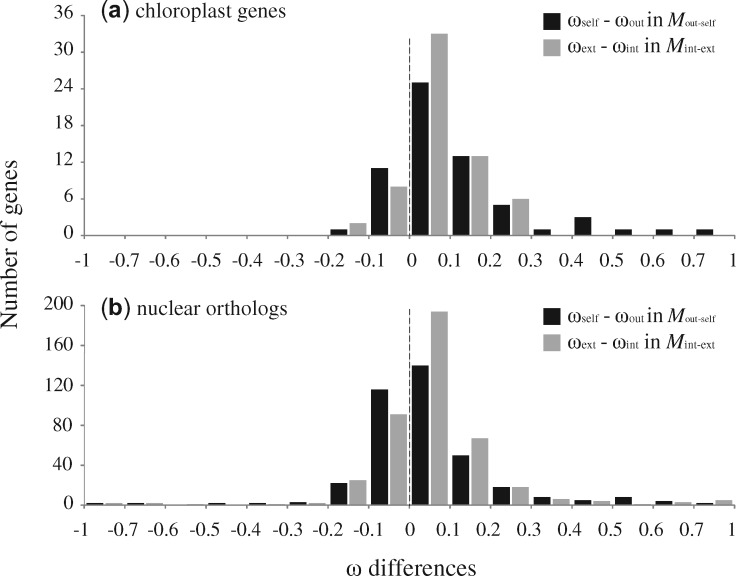
Distributions of the differences of ω (dN/dS) estimated separately in Mout-self and Mint-ext for (a) 72 chloroplast genes and for (b) 445 nuclear orthologs, respectively. Two ratios (ωout and ωself) in Mout-self are given for outcrossing and selfing branches. Two ratios (ωint and ωext) in Mint-ext are estimated for internal and external branches.
Molecular Evolution of the Nuclear Genome
We provide details of the de novo transcriptome assemblies and coding region identification and annotation in the supplementary results S1, Supplementary Material online. Values for genome-wide homozygosity of the homostylous species were ∼6-fold higher than for distylous species.
For the concatenated data set, both the two-ratio models Mout-self and Mint-ext provided a significantly better fit than either M0 (χ2=27.686, df=1, P < 0.001; supplementary table S2b, Supplementary Material online) or M3 (χ2=3.576, df=1, P = 0.059; supplementary table S2b, Supplementary Material online). The ω value was higher in selfing than outcrossing species (ωself=0.175 vs. ωout=0.157) in the model Mout-self, and ωext (0.179) was higher than ωint (0.146) in the model Mint-ext (supplementary table S2b, Supplementary Material online).
When analyses were performed on each gene independently, 41 orthologs (9.3% of the investigated genes) were identified as differentially evolving between distylous and homostylous species. Thirty (71%) showed a significantly higher ωself than ωout (fig. 2c). Few positively selected sites contributed to the elevated ωself in nuclear genes tested by the branch-site model (supplementary table S3, Supplementary Material online). Similarly, 78 orthologs (17.8%) revealed a significant difference between internal and external branches (fig. 2d): ωext was higher than ωint in 71 genes (92.3%). Similar to our results for chloroplast genes, we observed significantly higher values of dN/dS in selfing and external branches when these genes were analyzed independently (Wilcoxon-signed rank test, P < 0.001; fig. 3b).
Patterns of Polymorphism in Lineages of Primula chungensis with Contrasting Mating Systems
The chloroplast data set of the P. chungensis coding region (72 genes) contained a total of 62,766 sites, of which 16 and 10 were polymorphic in the outcrossing and selfing lineage, respectively. The two lineages shared no polymorphic sites in common and showed divergence at 20 sites. General patterns of nucleotide diversity variation between the two lineages were consistent with that of nuclear sequences described below (selfing vs. outcrossing for π: 0.007% vs. 0.010%; θW: 0.006% vs. 0.012%; table 1a). The πN/πS ratio was also higher in the selfing than outcrossing lineage (selfing vs. outcrossing: 0.385 vs. 0.368). The mean Tajima’s D was negative in the outcrossing lineage, but positive in the selfing lineage (table 1a).

| Lineage | n | Site Class | N sites | S | π (10−3) | θ W (10−3) | π N/πS | θ N/θS | D |
|---|---|---|---|---|---|---|---|---|---|
| (a) Chloroplast data | |||||||||
| Outcrossing lineage (distylous) | 5 | Synonymous | 14,549 | 7 | 0.19 | 0.23 | 0.368 | 0.391 | −1.222 |
| Nonsynonymous | 48,216 | 9 | 0.07 | 0.09 | |||||
| Selfing lineage (homostylous) | 8 | Synonymous | 14,547 | 4 | 0.13 | 0.11 | 0.385 | 0.455 | 0.826 |
| Nonsynonymous | 48,218 | 6 | 0.05 | 0.05 | |||||
| (b) Nuclear data | |||||||||
| Outcrossing lineage (distylous) | 4 | Synonymous | 435,550 | 1,321 | 1.65 | 1.64 | 0.280 | 0.287 | −0.050 |
| Nonsynonymous | 1,432,160 | 1,222 | 0.47 | 0.47 | |||||
| Selfing lineage (homostylous) | 9 | Synonymous | 435,534 | 1,331 | 1.17 | 1.12 | 0.284 | 0.295 | 0.131 |
| Nonsynonymous | 1,423,176 | 1,258 | 0.34 | 0.33 | |||||
Note.—Nucleotide diversity (π), Watterson’s theta (θW), and test for neutrality (D; Tajima’s D based on π and θW) for each lineage are given.
n, sample size; Nsites, number of sites; S, polymorphisms.
The nuclear genome data set of P. chungensis contained a total of 2,954 gene alignments and 1,858,710 sites. Compared with the outcrossing lineage (mean θW=0.074%, π = 0.074%; table 1b), the level of polymorphism of the selfing lineage was significantly reduced (mean θW=0.052%, π = 0.053%; table 1b). The ratio of nonsynonymous to synonymous nucleotide diversity (πN/πS) was slightly higher in the selfing than in the outcrossing lineage (0.284 and 0.280, respectively). The mean Tajima’s D was slightly negative for polymorphic sites in the outcrossing lineage but positive in the selfing lineage (table 1b), likely because of the higher subdivision among selfing populations, a result consistent with previous work on the phylogeography of the species (Zhou et al. 2017).
For the concatenated nuclear genomic data set of P. chungensis, we also conducted protein evolution analyses by adding three- and four-ratio models (M3-1 and M4; supplementary fig. S2, Supplementary Material online). Here, the M3-1 model, which allows different ω values for internal selfing, internal outcrossing, and external branches, was a better fit than model Mint-ext (χ2=7.788, df=1, P < 0.01; supplementary table S2c, Supplementary Material online). Significant divergence of the ω ratio was obtained between the two internal branches with the selfing lineage higher than outcrossing lineage (ωint-out=0.284; ωint-self=0.328; supplementary table S2c, Supplementary Material online).
McDonald–Kreitman Tests for Selection in Lineages of P. chungensis with Contrasting Mating Systems
We conducted McDonald–Kreitman (MK) tests on each nuclear locus in selfing and outcrossing lineages of P. chungensis and summarized our results using the neutrality index (NI). We found a significant mating system effect on the NI. The NI value in the outcrossing lineage was close to 1 (NI=1.079; Fisher’s exact test, P = 0.088; fig. 4a) which fit the null expectation of equal ratios of divergence and polymorphism. In contrast, there was a significant deviation of neutrality (NI=1) for the selfing lineage with higher NI=1.162 (Fisher’s exact test, P < 0.001; fig. 4a), and thus evidence for the accumulation of slightly harmful mutations.
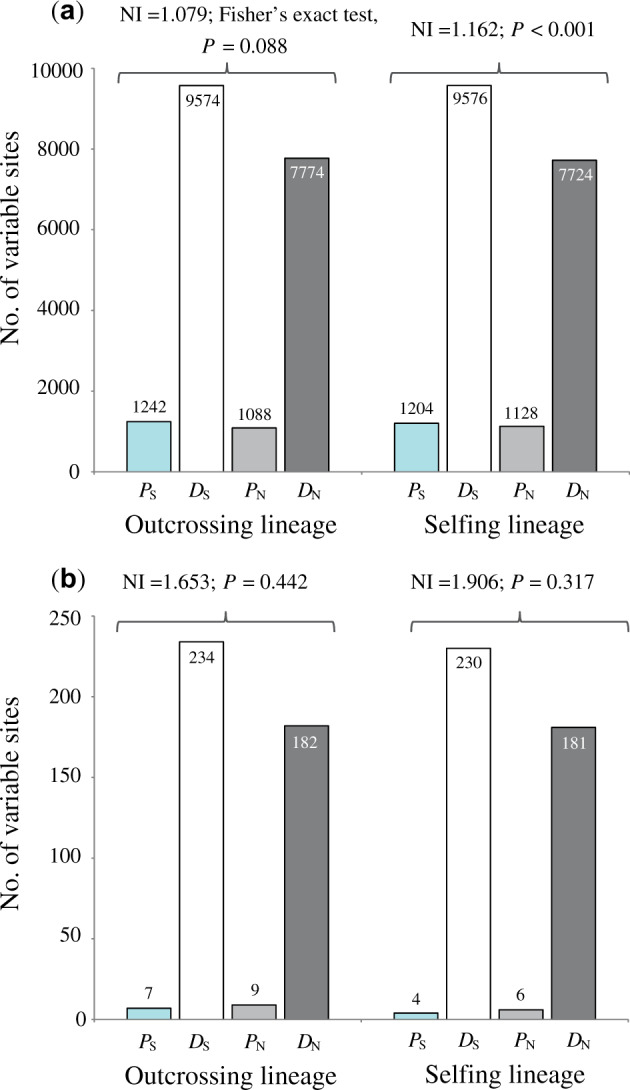
The MK test separating the outcrossing and selfing lineages of Primula chungensis from outgroup (P. mallophylla and P. secundiflora) based on (a) nuclear and (b) chloroplast genome data. Synonymous polymorphism (PS) and divergence (DS) are indicated by blue and white bars, respectively; nonsynonymous polymorphism (PN) and divergence (DN) are indicated by gray and black bars, respectively; numbers above each bar indicate the total numbers of each type of site.
We also conducted MK tests on the chloroplast data set of P. chungensis (fig. 4b). Both selfing (NI=1.906; Fisher’s exact test, P = 0.317) and outcrossing lineages (NI=1.653; Fisher’s exact test, P = 0.442) had higher NI values than 1, but the differences were not significant (fig. 4b).
Distribution of Fitness Effects of the Selfing and Outcrossing Lineage of P. chungensis
For the distylous and homostylous lineage of P. chungensis, we estimated the genome-wide distribution of fitness effects (DFE) of nonsynonymous mutations by using the folded synonymous and nonsynonymous site frequency spectra (SFS) (Eyre-Walker and Keightley 2009). We found differences between the outcrossing and selfing lineage (fig. 5). In the outcrossing lineage, there were relatively fewer effectively neutral nonsynonymous mutations, with ∼19% (SE=0.023) falling into this category (0 < Nes<1; fig. 5). In contrast, in the selfing lineage, ∼23% (SE=0.022) of new nonsynonymous mutations were classified as effectively neutral, and the difference between these proportions is significant (fig. 5). Furthermore, there was a significantly greater proportion of sites that were strongly selected against and very rarely fixed (Nes>100) in the selfing compared with the outcrossing lineage (45% vs. 24%; fig. 5).
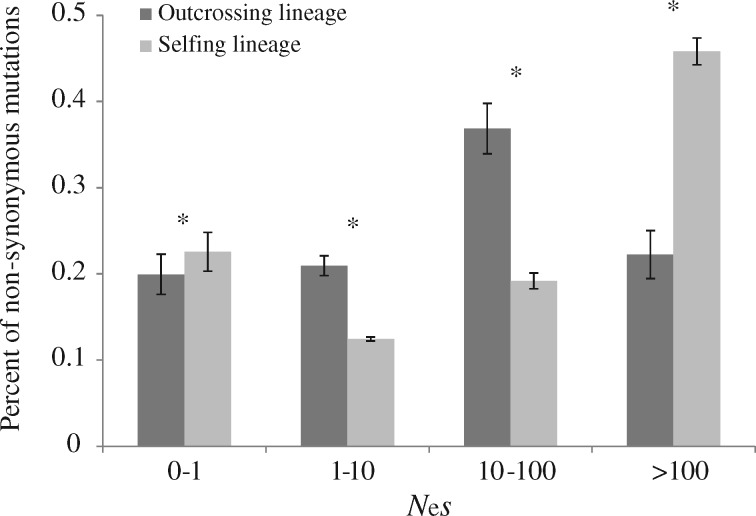
Distribution of fitness effects (DFE) of new nonsynonymous mutations falling in different Nes categories for outcrossing and selfing lineages of Primula chungensis. Nes is the product of Ne and the selection coefficient (s). Nine selfing and four outcrossing individuals were used to generate the DFEs. Error bars on top of each Nes category are 95% confidence intervals from 200 bootstrap replicates generated by resampling over loci. Comparisons between the two lineages at P < 0.05 significance level are indicated by *.
Adaptive Evolution of Selfing and Outcrossing Lineage of P. chungensis
We used the method of Eyre-Walker and Keightley (2009) to estimate the proportion of adaptive nonsynonymous substitutions (α) during the divergence of the selfing and outcrossing lineage of P. chungensis, with P. mallophylla as the outgroup. Our estimate of the proportion of adaptive substitutions differed significantly between the selfing and outcrossing lineage of P. chungensis (table 2). The estimate of α fixed by positive selection between the outcrossing lineage of P. chungensis and P. mallophylla was 25%, with the confidence interval (CI) not encompassing zero (95% CI: 5–44%). In contrast, no adaptive substitutions were found for the selfing lineage of P. chungensis since α was not significantly different from zero (95% CI: −16% to 32%) (table 2). Splitting nonsynonymous substitutions into adaptive (ωα) and nonadaptive proportions (ωd), we obtained a lower ωα and a higher ωd in the selfing compared with the outcrossing lineage of P. chungensis (table 2).
| Outcrossing Lineage | Selfing Lineage | |
|---|---|---|
| β (CI) | 0.322 (0.243,0.375) | 0.195 (0.166, 0.226) |
| Mean S (CI) | 69.048 (56.698, 97.609) | 732.82 (603.530, 915.048) |
| α (CI) | 0.250 (0.058, 0.445) | 0.163 (−0.167, 0.322) |
| ω α (CI) | 0.057 (0.012, 0.112) | 0.038 (−0.035, 0.079) |
| ω d (CI) | 0.170 (0.139, 0.231) | 0.198 (0.167, 0.247) |
β, the shape parameter of the gamma distribution assumed for the estimations of S.
Discussion
Using both chloroplast (72 genes) and nuclear coding sequences (445 orthologs), we found significantly higher nonsynonymous versus synonymous divergence ratios (dN/dS) in homostylous than distylous species (supplementary table S2, Supplementary Material online and fig. 3). This result likely reflects the fixation of deleterious mutations due to weaker selection over relatively long-time scales. Over shorter time scales, as evident in distylous and homostylous samples of P. chungensis (2,954 orthologs), weaker natural selection was also evident with a higher πN/πS ratio (table 1), a significantly different DFE (elevated proportion in: 0 < Nes<1; fig. 5), and a lower rate of adaptive evolution (α, ωα) in a selfing lineage than in an outcrossing lineage (table 2). These results are consistent with theoretical predictions of mating-system transitions from outcrossing to selfing at both macro- and microevolutionary time scales and are predicted features of the genomic selfing syndrome (Cutter 2019). Below, we compare our results with other recent genomic studies of selfing and outcrossing taxa and discuss their implications for the evolutionary fate of homostyles in Primula.
Mating Systems and Selection Efficacy
Contrasting patterns of selection are expected between related selfing and outcrossing populations for genes under strong purifying selection as indicated by a higher ratio of nonsynonymous to synonymous polymorphisms (πN/πS) and/or substitutions (dN/dS). In P. chungensis, we detected a modest signature of elevated πN/πS at both nuclear (0.284 vs. 0.280) and chloroplast genes (0.385 vs. 0.368) in our comparison of a selfing versus outcrossing lineage, respectively, a pattern consistent with the results of our MK tests (fig. 4). The reduced efficacy of selection in selfing samples was also evident by a significant increase in the proportion of effectively neutral nonsynonymous mutations (24% vs. 20% in the category: 0 < Nes<1; fig. 5). This result is in accord with DFE data from another heterostylous species, Eichhornia paniculata, in which the floral polymorphism has broken down and the transition to selfing is accompanied by a larger fraction of effective neutral sites, although in this case, the difference was not significant (Arunkumar et al. 2015).
One important caveat of our implementation of the DFE analysis is that this approach is likely to be biased when used in highly selfing populations because LD will result in per-locus estimates that are nonindependent of one another. Such an effect will depend critically on the amount of selfing in populations. Our previous SSR marker-based estimates (Zhong et al. 2019) indicated that the selfing rate of homostylous Primula populations averaged 0.8 (n = 15 populations) but was strongly associated with the degree of herkogamy (and see de Vos et al. 2018). Although we do not currently have selfing rate estimates for P. chungensis, the focal species in our DFE study, homostylous populations have relatively large flowers and a variable degree of stigma-anther separation and thus are unlikely to be highly selfing (Zhou et al. 2017). Therefore, our comparative use of the DFE in comparing distylous and homostylous population in P. chungensis is probably qualitatively valid, even if the point estimates we obtained are unlikely to be accurate.
In accord with the patterns we obtained for polymorphism data, the dN/dS ratio was also higher in homostylous than distylous species (supplementary table S2, Supplementary Material online). This result contrasts with several earlier studies using divergence data that have found no evidence of relaxed selection in selfing species (Wright et al. 2002; Cutter et al. 2008; Haudry et al. 2008; Escobar et al. 2010). In the case of homostylous Primula species, the transition to high selfing rates is usually rapid through an abrupt rearrangement of sexual organs in one generation (fig. 1a) and in some cases is likely associated with reproductive isolation and speciation, as appears to also be the case in Capsella (Mattila et al. 2018). This may help to explain why a signature of divergence between selfing and outcrossing lineages was also evident in P. chungensis (supplementary table S2c, Supplementary Material online).
Another issue that needs to be considered in detecting divergence differences between selfing and outcrossing lineages is the number of loci that are investigated. Insufficient genomic sampling probably accounts for the earlier finding of no signal of elevated dN/dS in a comparison homostylous versus distylous Primula species as only sequences from the plastid genes matK and rbcL were used (Glémin and Muyle 2014). In our study, genomic sampling involved the whole chloroplast genome, with 72 genes included in the analysis of protein evolution, and ω was significantly higher in homostylous than distylous branches (ωself>ωout in Mout-self, P < 0.001; fig. 3a). When we analyzed chloroplast genes independently, we identified 11 genes that evolved differentially between outcrossing and selfing species (fig. 2a). Although the chloroplast genes we analyzed are unlikely to evolve independently because of the absence of recombination, they are likely to mutate at different rates and the evolutionary fate of mutations at different genes will exhibit some degree of independence. Interestingly, matK did not show a significantly higher ωself than ωout (0.462 vs. 0.457, χ2=0.003, P = 0.959). And rbcL exhibited the opposite pattern to that predicted in Mself-out with ωself lower than ωout (0.057 vs. 0.149) and this model was rejected when compared with M0 (χ2=4.848, P = 0.028). Sample size issues were also apparent in studies of A. thaliana, with Slotte et al. (2010) finding evidence for weak purifying selection using genome-wide data in contrast to an earlier study by Wright et al. (2002) which failed to detect a signature using a more limited number of loci. Given the stochasticity and slow rate of mutation accumulation in chloroplast genes, large numbers of loci are probably required to consistently detect the influence of predominant selfing on plant genomes.
Although the genomic signatures we detected from polymorphism and divergence data are consistent with the expected effects of contrasting mating systems, the magnitude of the differences in ω values between homostylous and distylous species were relatively small (supplementary table S2, Supplementary Material online). A plausible explanation for this result is that we detected few positively selected sites in homostylous species (supplementary table S3, Supplementary Material online) but more efficient positive selection was evident in outcrossing lineages. As a result, adaptive substitutions should strongly increase the dN/dS ratio, and this in turn may lower the difference in dN/dS between outcrossing and selfing lineages. This explanation is supported by our analysis of genome-wide adaptive evolution which found a higher proportion of adaptive nonsynonymous substitutions (α) and adaptive rates (ωα) in distylous than homostylous lineage (table 2). Slower rates of adaptive evolution accompanying shifts in reproductive systems have also been reported in A. thaliana (Slotte et al. 2010), the selfing snail Galba truncatula (Burgarella et al. 2015), and asexual species of Oenothera (Hersch-Green et al. 2012).
Potential Implications for the Evolution of Homostyly in Primula
Our results provide insights on the evolutionary fate of homostylous Primula species, with potential implications for the evolution of selfing species in general. According to the “dead-end hypothesis” initially proposed by Stebbins (1957), selfing lineages are expected to arise frequently from outcrossing ancestors due to the transmission advantage of mating system modifiers and reproductive assurance under pollinator and/or mate limited conditions. But once selfers have originated they are more prone to extinction than outcrossers as a result of a variety of genetic and ecological factors (Busch and Delph 2012; Igic and Busch 2013; Wright et al. 2013; Barrett et al. 2014). Rapid transitions from outcrossing to selfing in Primula by single-gene (CYP734A50) mutations (Huu et al. 2016; Kappel et al. 2017) appear to be most commonly associated with speciation (cladogenic shifts) rather than species replacement (anagenic shifts) (see Goldberg and Igic 2012). However, over long-time scales, the frequent origin of homostyly in Primula (de Vos, Wüest, et al. 2014; Zhong et al. 2019) does not appear to contribute significantly to the overall species richness of the genus. Indeed, the “live fast and die young” nature of selfing homostylous lineages described for Primula helps to explain their transient nature over long-time scales (de Vos, Hughes, et al. 2014).
In most Primula species, enforced outcrossing, governed by heteromorphic self-incompatibility, may result in long-term genetic advantages by increasing opportunities for survival and adaptation. This may be of particular importance in the topographically complex mountains of southwest China where most species occur. In contrast, the negative genomic consequences of persistent selfing in homostyles, including reduced genetic diversity and the accumulation of deleterious mutations, may potentially limit long-term adaptive potential (although see Grant and Kalisz 2019). Thus, the evolution of the genomic selfing syndrome could be one of the primary drivers of the extinction of selfing lineages (Glémin and Ronfort 2013; Cutter 2019), despite the short-term advantages of selfing (Busch and Delph 2012) and ephemeral bursts of speciation (de Vos, Hughes, et al. 2014). Our empirical evidence of genomic degradation in homostyles of Primula is therefore generally consistent with Stebbin’s dead-end theory for selfing lineages.
Not all homostyles in heterostylous species necessarily have a doomed long-term fate. Several features of the reproductive and genetic systems of some taxa may ameliorate the negative effects of selfing and delay extinction risk. The simplest mechanism involves the evolution of some degree of herkogamy in homostyles resulting in the evolution of mixed mating rather than predominant selfing (Barrett and Shore 1987; de Vos et al. 2018). Investigations of species with mixed mating indicate that moderate levels of selfing are associated with more efficient selection and the absence of characteristic features of the genomic selfing syndrome (Salcedo et al. 2014; Laenen et al. 2018). Although we observed some evidence for the genomic syndrome in homostylous populations of P. chungensis, this species is unusual in displaying considerable variation in herkogamy thus providing opportunities for mixed mating (Zhou et al. 2017). Significantly, P. chungensis has a much larger geographical range than other homostyles from SW China that were investigated in this study. Similarly, three other genera (Dodecatheon, Cortusa, and Sredinskya), inferred to be descended from the most recent common ancestor of Primula (Mast et al. 2006) which was distylous, are each monomorphic with stigmas positioned above anthers (Mast and Reveal 2007). Their possession of approach herkogamy may represent a floral strategy that limits the most harmful genomic consequences of selfing when it arises through the breakdown of distyly.
A second mechanism that may reduce the extinction risk for some homostylous lineages of Primula is polyploidy. The ability to self may facilitate the establishments of polyploid and genome duplication can ameliorate the negative effects of selfing and inbreeding depression, particularly in autopolyploids (Rausch and Morgan 2005). A close association between polyploidy and homostyly is evident in Primula sect. Aleuritia (Guggisberg et al. 2006, 2009; Theodoridis et al. 2013). In this section, polyploid homostyles are more commonly found in previously glaciated areas of high latitude and appear to be more ecologically successful than their distylous ancestors (Guggisberg et al. 2006). However, there is no evidence that among the sections of Primula that are the basis of our study that polyploidy plays a significant role in the evolutionary fate of homostyles. Indeed, surveys of chromosome numbers indicate that the vast majority of the species we investigated are diploid, and the only known exception is P. oreodoxa which has both diploid and tetraploid homostylous populations (Yuan, Barrett, et al. 2017).
Conclusions
Our study represents the most comprehensive analysis of sequence divergence and polymorphism data based on both plastid and nuclear genes for related outcrossing and selfing plants. We found both macro- and microevolutionary evidence for components of the genomic selfing syndrome. The transition to selfing via homostyle evolution has resulted in significant effects on genetic diversity, the efficacy of selection, and on rates of adaptive evolution. Most studies on the genomic consequences of mating-system transition have focused on model systems by comparing related species pairs (e.g., Arabidopsis and Capsella, reviewed in Mattila et al. 2018) or populations (e.g., Eichhornia paniculata, Arunkumar et al. 2015) with contrasting mating systems. Primula provides a valuable opportunity to extend these approaches as it is composed of numerous independent transitions from outcrossing to selfing as a result of the repeated breakdown of distyly to homostyly. Future comparative investigations of transitions of different ages among Primula species should provide novel insights on diverse features of genome evolution and the extent to which the evolution of selfing increases extinction risk.
Materials and Methods
Plant Materials
To evaluate the potential influence of the evolutionary history of selfing on chloroplast sequence divergence, we obtained 56 samples each from 44 distylous and 12 homostylous species, and 22 samples from four distylous–homostylous species. We analyzed nuclear genome divergence by using the RNA-seq data of 12 samples each from nine distylous and three homostylous species. To investigate molecular divergence over shorter time scales, we focused on P. chungensis, a species with at least two independent origins of homostyly in the Tibet and Hengduan mountains region (Zhou et al. 2017). We sampled 26 individuals from 13 populations from the Hengduan mountains (Sichuan lineage); for population information see Zhou et al. (2017). Thirteen individuals were used for plastome assembly, with five and eight individuals each from distylous and homostylous populations, respectively. The remaining 13 individuals from four distylous and nine homostylous populations were investigated using RNA-seq. In total, we generated 51 new chloroplast genome assemblies and 18 transcriptomes with the chloroplast and RNA-seq data obtained from our previous phylogenomic study (Zhong et al. 2019) and Sequence Read Archives (see Zhang et al. 2013; Cocker et al. 2015; Nowak et al. 2015; Yuan, Zeng, et al. 2017), respectively. Information on all samples are presented in supplementary table S4, Supplementary Material online.
Chloroplast Genome Data Preparation for Molecular Evolutionary Analysis
We obtained chloroplast genomes of our samples (supplementary table S1, Supplementary Material online) from de novo assemblies following the procedures detailed below. First, we isolated total genomic DNA with a modified cetyl trimethyl ammonium bromide method (Doyle 1991) from silica-dried leaf tissue. Then we constructed libraries using a NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA). Paired-end sequencing was carried out on a HiSeq X-Ten sequencer (Illumina, San Diego, CA) with 2 × 150 bp read length. We obtained ∼2 Gb of sequence data for each sample. After quality control using NGS QC TOOL Kit (Patel and Jain 2012) with default parameters, we performed de novo assemblies of chloroplast genomes employing the GetOrganelle pipeline (Jin et al. 2018) for each accession. We subsequently annotated the circular plastomes following the PGA program (Qu et al. 2019) and all the annotated transfer RNA (tRNA) genes were further verified using the corresponding structures predicted by tRNAscan-Se v.1.21 (Schattner et al. 2005). We retrieved five annotated Primula plastid genomes from NCBI database to complement our data set. Finally, we extracted 72 chloroplast coding genes from each annotated plastome for further molecular evolutionary analysis.
Transcriptome Data Preparation for Molecular Evolutionary Analysis
We isolated total RNA of each sample using an RNAprep Pure Plant Kit (TIANGEN Biotechnologies Corporation, Beijing, China). We prepared the cDNA library for transcriptome sequencing using a cDNA Synthesis Kit (Illumina, San Diego, CA) following the manufacturer’s recommendations, and the 18 cDNA libraries were then sequenced on the Illumina HiSeq 4000 platform (Illumina, San Diego, CA) to obtain short sequences of 150 bp from both ends of each cDNA.
We performed preliminary quality check of raw reads within each data set with FastQC v.0.11.2. The clean reads were filtered from the raw reads with Trimmomatic v.0.32 (Bolger et al. 2014) after trimming adapters and removing both ambiguous (N>10%) and low-quality reads (Phred score<30). We generated de novo assemblies of transcriptomes of individuals for each species using the Trinity platform with default parameters (Haas et al. 2013). We discarded contigs with length <300 bp due to their low annotation rate. To remove the redundant sets of sequences, similar sequence stretches were filtered and merged with the TGI Clustering tool (Pertea et al. 2003). We selected the longest transcript as representative for each cluster. We characterized the quality of assemblies, by examining the RNA-seq read representation by aligning reads to the assemblies using bowtie2 (Langmead and Salzberg 2012) and counted the overall alignment rate and number of proper pairs. We performed an assessment of the representation of single-copy conserved plant orthologs using the program BUSCO (Simão et al. 2015) to evaluate whether the assembly was full-length or nearly full-length. We calculated the ExN50 statistic against a fraction of the most highly expressed transcripts (Ex), and retained transcripts with an expression level higher than the saturation point for subsequent analysis.
We predicted open reading frames (ORFs) from the assembled transcripts using the program TransDecoder and scanned the predicted ORFs for homology to known proteins in two ways: a BLAST search (Camacho et al. 2009) against Swiss-Prot database, and by identifying hidden Markov model protein domains with HMMER v.3.1b (Eddy 2011) against the HMMER/Pfam protein database. Those coding peptides with BLAST hits (e-value<1e−5) or domain hits (FullSeqScore and FullDomainScore>20) were retained in the set of reported likely coding regions. We integrated the final coding region predictions BLAST and Pfam search results using TransDecoder. We selected the top scores for predicted ORFs for each transcript in situations where there was more than one prediction within a transcript.
Finally, we used OrthoFinder v.2.3 (Emms and Kelly 2019) to identify 445 single-copy orthologs among annotated ORFs data set in the 12 Primula species for interspecific divergence analysis, and 2,954 orthologs from the 13 individuals of P. chungensis used for intraspecific divergence and polymorphism analysis. We aligned the protein-coding sequences corresponding to orthologous genes with ParaAT (Zhang et al. 2012). We excluded misalignments manually based on the sequence alignment for each ortholog set.
Molecular Divergence between Distylous and Homostylous Species
To investigate the association between mating-system differentiation and the accumulation of deleterious mutations, we estimated the ratio of nonsynonymous to synonymous divergence (dN/dS=ω) among distylous and homostylous species. We used the codeml program, included in PAML package v.4.4 (Yang 2007), to test the fit of several hierarchical models using both plastid and nuclear sequences. We evaluated the fit of the following maximum likelihood branch models to the data: 1) the null one-ratio model (M0), which assumes the same ω value for all branches; 2) a two-ratio model, in which two separate ω values were estimated for distylous and homostylous species (Mout-self; fig. 1b); 3) a second two-ratio model contrasting internal versus external branches (Mint-ext; fig. 1c); 4) a three-ratio model (M3; fig. 1d), where internal and external distylous and external homostylous branches were allowed to have different ω. We applied different models to both individual gene alignments and to all genes from concatenated data sets. We used the branch-site model to test if there was any evidence for signatures of positive selection of loci with significantly elevated ω values in homostyles. We then compared the fit of models by using likelihood ratio tests with the appropriate degrees of freedom (df).
For the chloroplast data set, we estimated codon evolution based on the phylogenetic tree reconstructed by RAxML v.8 (Stamatakis 2006) with 1,000 bootstrap under the GTRGAMMA model (supplementary fig. S1, Supplementary Material online), and the outgroup Androsace bulleyana (accession number: KU513438) was excluded from the tree in the PAML analyses. For the transcriptome data set, we examined the inter- and intraspecific analyses separately using the inferred phylogenetic tree specific to the corresponding inter- and intraspecific orthologs.
Within-Species Polymorphism in Distylous and Homostylous Lineages of P. chungensis
To test the hypothesis that homostylous populations of P. chungensis have lower genetic diversity and a higher ratio of nonsynonymous versus synonymous nucleotide diversity (πN/πS) compared with distylous populations, we estimated the population statistics of nucleotide diversity (π), Watterson’s theta (θW), and Tajima’s D for both chloroplast and nuclear sequences. We then used a contingency table-based variation of the MK test (McDonald and Kreitman 1991) and Fisher’s exact test to evaluated whether the ratio of nonsynonymous to synonymous polymorphisms (PN/PS) of P. chungensis was differentiated from the ratio of nonsynonymous to synonymous divergence (DN/DS) of the outgroup species (P. secundiflora and P. mallophylla, both distylous). We summarized the MK results using the NI, defined as (PN/DN)/(PS/DS) (Rand and Kann 1996).
We also performed tests of selection on distylous and homostylous population samples of P. chungensis by estimating the genome-wide DFE of new nonsynonymous mutations, the rate of adaptive evolution (α), and the rate of adaptive [ωα=α (dN/dS)] and nonadaptive [ωd=(1−α) (dN/dS)] nonsynonymous mutations using DFE-α v.2.15 (Eyre-Walker and Keightley 2009). For these analyses, synonymous sites were assumed to evolve neutrally and we obtained the estimates of DFE and α for both selfing and outcrossing lineages. We generated the locus-specific nonsynonymous- and synonymous-folded SFS and number of invariant sites using a Perl script, Polymorphorama (Bachtrog and Andolfatto 2006) and we report the proportion of mutations falling into a given Nes range (<1, 1–10, 10–100, and >100) of the DFE. We estimated the rate of adaptive evolution (α) by comparing the selfing and outcrossing lineages of P. chungensis with the outgroup P. mallophylla. We derived CIs and SEs from 200 bootstrap replicates by randomly resampling across loci using R (R Development Core Team 2014).
Acknowledgments
We thank Asher Cutter, Ramesh Arunkumar, and Stephen Wright for helpful comments on the article. This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31000000), the National Natural Science Foundation of China (31971394, 31770417, and 31570384), the Yunling Scholarship of Yunnan Province (YLXL20170001), Light of West China Program of the Chinese Academy of Sciences, and a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada.
Author Contributions
W.Z., S.C.H.B., H.W., and D.-Z.L. planned and designed the research. X.-J.W., W.Z., L.Z., and Z.-K.W. performed experiments and conducted field work. X.-J.W. and W.Z. analyzed the data. W.Z., X.-J.W., and S.C.H.B. wrote the article.