 Recent trends in molecular diagnostics of yeast infections: from PCR to NGS
Recent trends in molecular diagnostics of yeast infections: from PCR to NGS
FEMS Microbiology Reviews


- Altmetric
- INTRODUCTION
- MOLECULAR IDENTIFICATION OF TARGETED DNA REGIONS
- SPECTROSCOPY-BASED METHODS
- ANTIBODY-BASED TECHNIQUES
- ANTIFUNGAL SUSCEPTIBILITY TESTING
- DIAGNOSIS OF INFECTION TYPE AND STAGE
- TRENDS IN NGS
- BIOINFORMATICS TOOLS AND DATABASES RELEVANT FOR DIAGNOSTICS OF YEAST PATHOGENS
- FUTURE PROSPECTS AND CONCLUDING REMARKS
- COLLABORATORS
- Supplementary Material
- FUNDING
The incidence of opportunistic yeast infections in humans has been increasing over recent years. These infections are difficult to treat and diagnose, in part due to the large number and broad diversity of species that can underlie the infection. In addition, resistance to one or several antifungal drugs in infecting strains is increasingly being reported, severely limiting therapeutic options and showcasing the need for rapid detection of the infecting agent and its drug susceptibility profile. Current methods for species and resistance identification lack satisfactory sensitivity and specificity, and often require prior culturing of the infecting agent, which delays diagnosis. Recently developed high-throughput technologies such as next generation sequencing or proteomics are opening completely new avenues for more sensitive, accurate and fast diagnosis of yeast pathogens. These approaches are the focus of intensive research, but translation into the clinics requires overcoming important challenges. In this review, we provide an overview of existing and recently emerged approaches that can be used in the identification of yeast pathogens and their drug resistance profiles. Throughout the text we highlight the advantages and disadvantages of each methodology and discuss the most promising developments in their path from bench to bedside.
The authors discuss the current status of the use of high-throughput (-omics) technologies on the diagnostics of yeast infections.
INTRODUCTION
Opportunistic yeast pathogens cause a wide range of superficial to systemic infections, which can often be fatal (Kullberg and Arendrup 2015). The incidence of these pathogens has increased in recent years, becoming a major source of life-threatening nosocomial infections. This is partly due to medical progress, which results in increased survival of particularly susceptible patients, such as premature neonates, elderly people and immunocompromised patients. In addition, the extensive use of catheters, broad-spectrum antibiotics and abdominal surgery favors the spread of opportunistic yeasts from their normal commensal niches (Turner and Butler 2014). Among pathogenic yeasts, Candida spp. are the most common cause of threatening invasive infections (Brown et al. 2012). Oral/esophageal, vulvovaginal, bloodstream and intra-abdominal infections caused by Candida spp. (i.e. candidiasis) have an estimated annual incidence of ∼2.3 million, ∼134 million, ∼650 000 and ∼80 000 cases, respectively (Levallois et al. 2012; Sipsas and Kontoyiannis 2012; Bassetti et al. 2013; Bongomin et al. 2017; Pieralli et al. 2017). Despite recent advances, the mortality rates associated with invasive candidiasis remain high at around 40%, and their treatment is complicated by increasing resistance to antifungals, as well as the appearance of novel pathogenic species (Papon et al. 2013; Gabaldón, Naranjo-Ortíz and Marcet-Houben 2016). Although the most common cause of candidiasis is Candida albicans, the emergence of non-albicans Candida species such as Candida dubliniensis, Candida glabrata, Pichia kudriavzevii (syn. Candida krusei (Douglass et al. 2018)), Candida parapsilosis and Candida tropicalis has increased over the past decades (da Matta, Souza and Colombo 2017), and Candida auris has recently been recognized as a globally emerging multidrug-resistant species (Geddes-McAlister and Shapiro 2018; Sekyere and Asante 2018). Currently, over 30 different Candida spp. have been identified as causative agents of candidiasis (Papon et al. 2013; Gabaldón, Naranjo-Ortíz and Marcet-Houben 2016). Furthermore, hybridization among pathogenic and non-pathogenic lineages can give rise to new virulent ones (Mixão and Gabaldón 2018). Candida spp. do not belong to a single genus in the phylogenetic sense, as different Candida species are spread throughout the Saccharomycotina tree (Kurtzman, Fell and Boekhout 2011; Gabaldón, Naranjo-Ortíz and Marcet-Houben 2016). Although for convenience the name Candida is still widely used in the clinical setting, it is important to understand that the term encompasses a wide diversity of species that display important differences in terms of clinically relevant phenotypes. We anticipate that current efforts in the field of yeast genomics and taxonomy will result in renaming of many of the clinically relevant Candida species, and it would be advisable for clinicians to be prepared for this change. From a diagnostics perspective, providing resolution at the species level (or even beyond) is important to guide therapy, because virulence and antifungal resistance vary between species (Schmalreck et al. 2014), and even between strains of the same species (Farmakiotis and Kontoyiannis 2017). Therefore, specific, accurate and fast diagnosis of the causative agent of infections is crucial in order to rapidly start appropriate antifungal therapy, especially in patients suffering from life-threatening candidiasis.
Classical diagnosis of candidiasis is based on microscopy, selective culture and/or biochemical approaches (Ellepola and Morrison 2005; Cuenca-Estrella et al. 2012; Arendrup et al. 2014a). All these methods require isolation and cultivation of the infective agent from clinical specimens, a process that takes around 48 h for most common pathogenic yeasts, and can require more time for some samples or species. In addition, the identification procedures require specific expertise, may provide ambiguous results and are generally time-consuming, causing further delay to an effective diagnosis. For this reason, there is a growing interest in the development of alternative methods, based on direct detection of diagnostic molecules. These approaches, collectively referred to as molecular diagnostics, have the potential to be directly applied to clinical specimens, and include proteomics-based methods and the detection of specific DNA sequences. Available methods and those currently under development differ in the need for cultivation of the infectious agent, the ability to directly use a clinical sample, sensitivity and accuracy, cost, time and expertise requirements, as well as in the range of species that can be identified. In addition, some emerging methods hold the promise of being able to readily diagnose both the species and drug resistance profile of the infecting agent. A common drawback of methods based on the detection of DNA is that the detection of the DNA may not necessarily correlate with the presence of actively infecting cells (i.e. if the species can also be a commensal), or even with the presence of living cells (DNA from dead cells can also be detected). For this reason, several recent approaches are based on the detection of RNA from actively transcribed genes, which are a better proxy for active cells, and which may also reveal signatures that distinguish invasive from commensal behaviors.
The field of diagnosis of yeast infections has advanced significantly in the last decade and is currently experiencing a revolution with the advent of novel sequencing and proteomics technologies. Yet, there is a long way to go from the successful proof of concept of a novel diagnostic method to its readiness for routine clinical use. Ideally, diagnostic tools must be cheap, fast, sensitive, accurate and easy to use. Moreover, they should aim to identify a broad spectrum of species. Currently, several molecular-based diagnostic tools for yeasts are commercially available. However, they focus on the main pathogenic species, while the detection of rare and emerging yeast pathogenic species is lagging behind. This was emphasized by the unprecedented outbreaks of drug-resistant isolates of C. auris in hospital environments that were initially misidentified by available commercial systems (Kathuria et al. 2015). In this review, we provide a comprehensive survey of existing approaches that are used in the identification of yeast pathogens and their drug resistance profiles. Throughout the review, we particularly emphasize the advantages and disadvantages of each approach and highlight the most promising recent developments brought about by emerging technologies. Figure 1 and Table 1 provide an overall summary of the main available approaches and methods.

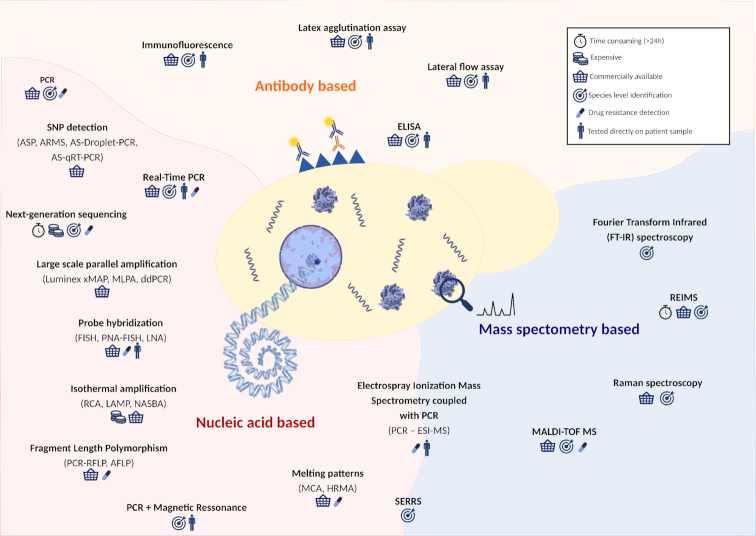
Overview approaches for the detection of fungal pathogens. Schematic representation of the different technologies used for the identification of fungal organisms. These techniques can be based on mass spectrometry (blue background), nucleic acid (red background) or antibody (orange background). Techniques based on more than one of these aspects are represented in the border of the respective divisions. ASP—allele-specific PCR; ARMS—amplification refractory mutation system; AS-Droplet-PCR—combination of ASP with droplet PCR; AS-qRT-PCR—combination of ASP with quantitative PCR; MLPA—multiplex ligation-dependent probe amplification; ddPCR—droplet digital PCR; FISH—fluorescent in situ hybridization; PNA-FISH—peptide nucleic acids-FISH; LNA—locked nucleic acids; RCA—rolling-circle amplification; LAMP—loop-mediated isothermal amplification; NASBA—nucleic acid sequence-based amplification; RFLP—restriction fragment length polymorphism; AFLP—amplified fragment length polymorphism; ELISA—enzyme-linked immunosorbent assay; MALDI-TOF MS—matrix-assisted laser desorption-time of flight mass spectrometry; PCR-ESI-MS—electrospray ionization mass spectrometry coupled with broad-spectrum PCR; SERRS—surface-enhanced resonance Raman spectroscopy; MCA—melting curve analysis; HRMA—high-resolution melting analysis.

| Assay (manufacturer) | Biomarker—principle | Scope | Goal | Sensitivity/specificity (%) | Approval | LoD* | Time | Clinical specimens | References |
|---|---|---|---|---|---|---|---|---|---|
| ePlex BCID-FP (GenMark DX) | DNA—r.f.u. | 19 Candida spp. | I | u.d./u.d. | u.d. | — | 1.5 h | Blood culture | Maubon et al. 2018 |
| Yeast Traffic Light PNA FISH™ (AdvanDX) | DNA—r.f.u. | 5 Candida spp. | I | –/– | FDA | — | 1.5 h | Blood culture (+) | Ibáñez-Martínez, Ruiz-Gaitán and Pemán-García 2017 |
| FilmArray® BCID Panel (Biomerieux) | DNA—r.f.u. | 5 Candida spp. | I + R | 100/99.8–100 | IVD/CE | — | 1 h | Blood culture | Southern et al. 2015; McCoy et al. 2016; Salimnia et al. 2016 |
| T2 Candida Panel (T2 Biosystems) | DNA—r.f.u. (MR) | 5 Candida spp. | I | 91.1/99.4 | FDA | 1 cfu/mL | 4.5 h | Blood | Mylonakis et al. 2015; Pappas et al. 2016; Pfaller, Wolk and Lowery 2016; Zervou et al. 2017 |
| IRIDICA BAC BSI (Abbott Diagnostics) | DNA—PCR + MS | Pan-microbial | I | 81/84 | IVD/CE/FDA | 8 cfu/mL | 6 h | Blood, tissue, OSF, BAL, ETA | Metzgar et al. 2016; Stevenson et al. 2016 |
| UMD Universal (Molzym) | DNA—ITS-based qPCR + seq | Pan-fungal + pan-bacterial | I | –/– | IVD/CE | 10 cfu/mL | 24 h | Body fluids, tissue, swabs | Kühn et al. 2011; Borde et al. 2015 |
| SeptiTestTM (Molzym) | DNA—qPCR + seq | Pan-fungal + pan-bacterial | I | 48/86 | IVD/CE | — | 7–8 h | Blood | Stevenson et al. 2016 |
| Light Cycler SeptiFast Test MGRADE® (Roche) | DNA—LC multiplex PCR | 5 Candida spp. | RI | 65/86 | IVD/CE | 30 cfu/mL | 6 h | Blood | Stevenson et al. 2016 |
| Prove-it®Sepsis (MobiDiag) | DNA—PCR + hyb | Pan-yeast (incl. 8 Candida spp.) | I + R | 95/99 | IVD/CE | — | 3.5a h | Blood | Aittakorpi et al. 2012 |
| MagiplexTM Sepsis Real-time Test (Seegene) | DNA—multiplex qPCR | Sepsis panel (incl. 5 Candida spp.) | D + I | –/– | IVD/CE | — | 3a h | Blood | Denina et al. 2016 |
| RenDx Fungiplex (Renshaw Diagnostics) | DNA–28S rRNA PCR-SERS | Pan-Aspergillus + pan-Candida | D + I | 80 (Candida)/87.5 | IVD/CE | 20 g/r | 6 h | Blood | White et al. 2014 |
| MycoReal Candida (Ingenetix) | DNA—ITS2 PCR LC-hyb | 7 Candida spp. | I | –/– | RUO | — | 2a h | n.s. | Mutschlechner et al. 2016 |
| RealLine (Bioron) | DNA—multiplex STI Kits | C. albicans + Gardnerella vaginalis | D + I | –/100 | IVD/CE | — | 2 h | Saliva, urine, SEC | Bioron 2016 |
| AllplexTM STI/BV Panel Assays-Candidiasis Assay (Seegene) | DNA—multiplex qPCR | STI panel (incl. 7 Candida spp.) | D + I | –/– | IVD/CE | — | 2.5a h | Genital swab, urine, LBCS | Seegene 2019 |
| FungiXpert (Era Biology) | DNA—multiplex qPCR | C. albicans/Aspergillus | D + I | –/– | IVD/CE | — | 2a h | BAL, fluids | Era Biology 2018 |
| AmpliSens® Candida albicans-FEP (AmpliSens®) | DNA—qPCR | C. albicans | I | –/– | IVD/CE | 1000– 2000 cfu/mL | — | Urogentital swabs and urine samples | AmpliSens®2018 |
| Fungiplex® Aspergillus Azole-R IVD Real-Time PCR Kit (Bruker Daltonics) | DNA—multiplex qPCR | Mutation in Cyp51A (TR34 and TR46) | DI | 100/(LoD = 50 ge) 100 | IVD | 50 ge | <2 h | Serum, plasma and BAL | Bruker 2018a |
| Fungiplex® Universal RuO Real-Time PCR Kit (Bruker Daltoics) | DNA—multiplex qPCR | All fungal DNA plus internal control | BI | 98/98 | RUO | 50 input copies | <2 h | n.s. | Bruker 2018b |
| Fungiplex® Candida auris Real-Time PCR Kit (Bruker Daltonics) | DNA—qPCR | C. auris plus internal control | DI | 97.3/99.8 | RUO | 10 input copies | <2 h | n.s. | Bruker 2018c |
| Fungitell (Associates of Cape Cod) | Antigen–1,3-β-d-glucan detection | Pan-fungal (except Mucormycetes) | D | depends on cohort/81.1–86.5 | IVD/CE/FDA | n/a | 1 h | Serum, CSF | Liss et al. 2016; Stevens et al. 2016 |
| Platelia Candida Ag Plus (BioRad) | Antigen—Candida mannan detection by ELISA | Candida spp. | D | 40–70%sd/70–98 % | IVD/CE | n/a | 4 h | Serum, plasma | Lunel et al. 2011 |
| Candida antigen Serion ELISA (Institut Virion\Serion GmbH) | Antigen—Candida mannan detection by ELISA | Candida spp. | D | 52/98 | IVD/CE | n/a | 4 h | Serum, plasma | Hartl et al. 2018 |
| Cryptococcal Antigen Lateral Flow Assay (IMMY) | Antigen—IC detection | Cr. neoformans/Cr. gattii | D | 98–100/97–100 | IVD/CE/FDA | n/a | 15 min | Serum, CSF | Vidal and Boulware 2015 |
| Cryptococcal Antigen Latex Agglutination Assay (IMMY) | Antigen—Latex agglutination | Cr. neoformans | D | 93–100/93–100 | IVD/CE | 0.5–25 ng/mL | 2 h | Serum, CSF | Binnicker et al. 2012 |
| Cryptococcal Antigen enzyme immunoassay (IMMY) | Antigen—ELISA | Cr. neoformans/Cr. gattii | D | 98r/97r | IVD/CE | n/a | 3 h | Serum, CSF | Binnicker et al. 2012 |
| Histoplasma Antigen EIA (IMMY) | Antigen—ELISA | Histoplasma capsulatum | D | 64/99 | IVD/CE | n/a | 4 h | Urine | LeMonte et al. 2007; Theel et al. 2013 |
| Cryptococcal Antigen Latex Agglutination System (Meridian Bioscience) | Antigen—Latex agglutination | Cr. neoformans | D | 100/100 | IVD/CE | n/a | 4.5 h | Serum, CSF | Jaye et al. 1998 |
| Premier® Cryptococcal Antigen (Meridian Bioscience) | Antigen—ELISA | Cr. neoformans | D | 100/97 | IVD/CE | n/a | 1.5 h | Serum, CSF | Gade et al. 1991 |
| IR Biotyper Kit (Bruker Daltonics) | Polysaccharides—r.f.u. | All microorganisms | ST | –/– | RUO | — | <3 h | Agar plate | Bruker 2018d; Bruker 2018e |
| MALDI Sepsityper Kit (Bruker Daltonics) | Protein—r.f.u. (MALDI-TOF MS) | Yeasts + bacteria | I | –/– | IVD/CE | 1 mL | 15–20 min | Blood culture (+) | Scohy et al. 2018 |
| AuxaColor™ 2 (Bio-Rad) | Enzymatic—color pH indicator | 33 most frequently isolated yeasts in medical mycology | I | –/– | FDA | — | 24–48 h | Blood culture (+) | Ibáñez-Martínez, Ruiz-Gaitán and Pemán-García 2017 |
| BactiCard® Candida (Remel, Lenexa, KS, USA) | Enzymatic—using NGL/PRO | Mainly C. albicans | I | 97.8/92.5 | IVD | — | 30 s | Agar plate | Cárdenes et al. 2004 |
| Candida albicans Test Kit (MUREX C. albicans 50) (Remel) | Enzymatic—using NGL/PRO | Mainly C. albicans | I | 100/100 | IVD | — | 0.5–1 h | Agar plate | Fenn et al. 1996 |
| O.B.I.S. C. albicans(Oxoid) | Enzymatic—using NGL/PRO | Mainly C. albicans | I | 100/100 | — | 3 to 5 colonies (each 1 mm) | 0.5–1 h | Agar plate |
r.f.u.—ready for use; MR—magnetic resonance; MS—mass spectrometry; seq—sequencing; LC—light cycler (Roche); hyb—hybridization; STI—sexually transmitted infection; IC—immunochromatographic; NGL—β-galactosaminidase; PRO—proline arylamidase; u.d.—under development; FDA—The Food and Drug Administration, USA; IVD—in vitro diagnostics; CE—European conformity; RUO—research use only; sd—highly species dependent; r—retrospective; *—for Candida; g/r—genomes/reaction; ge—genome equivalents; n/a—not available; a—excluding DNA extraction; (+)—positive; OSF—other sterile fluids; BAL—bronchoalveolar lavage; ETA—endotracheal aspirate; n.s.—not specified; SEC—scrapings of epithelial cells; LBCS—liquid-based cytology specimen; CSF—cerebrospinal fluid; I—identification; D—detection; RI—rapid identification; DI—direct identification (without culturing); BI—broad identification (without culturing); D + I—detection and identification; R—detection of resistance; ST = strains sub-typing. The authors to not claim to be complete, additional Candida detecting system might have been marketed with EC, IVD or FDA label.
MOLECULAR IDENTIFICATION OF TARGETED DNA REGIONS
Polymerase chain reaction (PCR) enables the selective amplification of a targeted segment of DNA, generating millions of copies of that sequence (amplicon) within a few hours (Mullis et al. 1986). The potential diagnostic use of this technique is obvious, as it allows the selective detection of minute amounts of the target DNA by using specific oligonucleotides. Diagnosis can be based merely on the presence of the amplicon (if it is unique for the targeted species), its particular size or its specific sequence, which can be determined by sequencing or by hybridization to a specific probe. The combination of specific PCR designs with subsequent analysis has led to a plethora of alternative PCR-based approaches that are increasingly used in the diagnosis of yeasts infections (Fig. 2). In addition, specific patterns in the DNA of infectious microorganisms can be detected without the need for selective amplification by PCR, for instance by means of direct hybridization with specific probes or by recognizing patterns in the length of fragments produced by enzymatic digestion of the DNA by specific endonucleases. These approaches will also be discussed in this section.
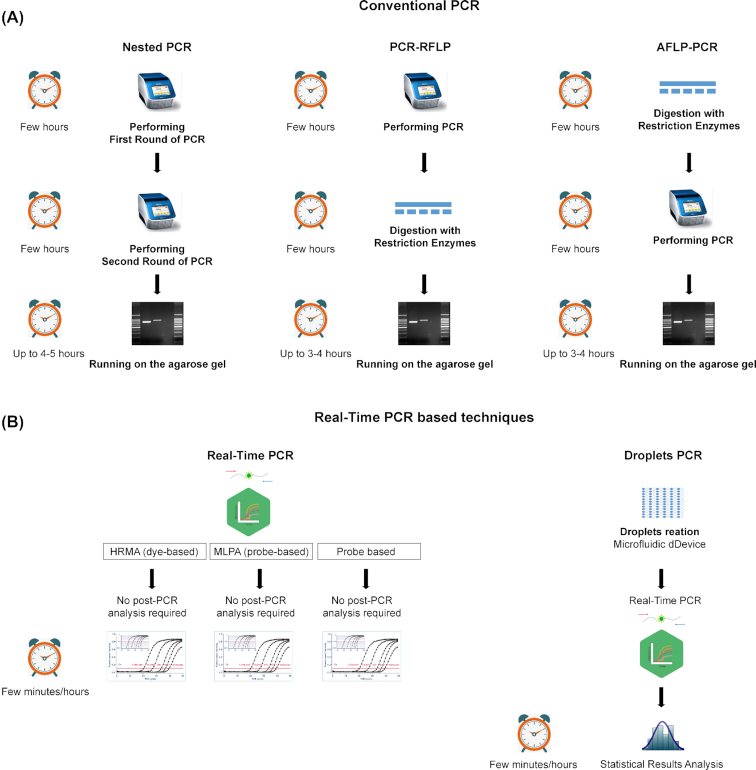
Scheme of commonly used PCR-based approaches used for fungal diagnostics. (A) Conventional PCR-based methods and (B) Real-time PCR-based methods used in diagnostics. For every method the intermediate steps are shown in a vertical manner. Next to every method the turn-around time is estimated from hours to minutes. RFLP—restriction fragment length polymorphism; AFLP—amplified fragment length polymorphism; HRMA—high-resolution melting analysis; MLPA—multiplex ligation-dependent probe amplification.
End-point PCR-based amplification
End-point PCR is routinely used for the detection and identification of specific infectious agents from cultures or directly from clinical specimens (Kourkoumpetis et al. 2012). Traditionally, detection and identification of pathogens is based on the development of primer sets, which selectively amplify a target locus. The locus can be species-specific, rendering an amplicon only if the target species is present, or have a broader spectrum, producing an amplicon from several species. In the latter case, differences in length, melting temperature or sequence between the amplicons can allow a more specific identification. The ribosomal RNA gene (rDNA), which is conserved and present in multiple copies, has been largely used as a target locus (Kurtzman and Robnett 1997). This locus presents a series of features that makes it particularly suited for diagnostics, in particular its presence in multiple copies, which allows amplification even from few cells, and its intrinsically high level of variation present in some regions, which allows the development of species-specific assays. The amplification and sequencing of the internal transcribed spacer (ITS) of the rDNA locus was agreed to be the international gold standard for the identification of fungal species (Schoch et al. 2012; Irinyi et al. 2016), and several universal primers do exist that amplify this region (see supplementary Fig. S1 available online). However, other regions of the rDNA locus can be more informative for certain clades or species, such as the Intergenic Spacer 1 (IGS1) region for Trichosporum (Sugita et al. 2002). In addition, other markers such as beta-tubulin or translation elongation factor genes can also be used in other fungal species (Irinyi et al. 2016). Nowadays with the vast development of bioinformatics and the availability of whole-genome sequencing data, the identification of species-specific or diagnostic regions is easier, faster and can provide higher specificity (Bohle and Gabaldón 2012; Capella-Gutierrez, Kauff and Gabaldón 2014).
Species-specific PCR primers for common fungal species belonging to the major human pathogenic genera, such as Candida,Aspergillus,Cryptococcus and Pneumocystis, are more commonly used in routine laboratories than broad-spectrum primers. However, a disadvantage is the lack of species-specific commercial assays for less common species within those genera, as well as for other emerging fungal genera that often cause severe and fast-progressing infections (Lackner, Caramalho and Lass-Flörl 2014). The advantage of pan-fungal or broad-spectrum PCR primers is that they allow the detection of both common and rare fungi. However, disadvantages include the fact that the results need to be interpreted by experts, since commonly occurring, non-pathogenic, commensal or saprotrophic fungi can yield a positive result due to the sensitivity of the assay (Lackner and Lass-Flörl 2017). A recently developed multiplex panel (YEASTpanel) is able to detect 21 clinically important yeast species belonging to the genera Candida,Trichosporon,Rhodotorula,Cryptococcus and Geotrichum, which collectively represent 95% of yeast infections (Arastehfar et al. 2019). A list of selected examples of commercially available pan-microbial, pan-fungal and Candida-specific kits is provided in Table 1. In many cases, sequencing of the amplicon is required to provide a specific diagnosis. The potential of PCR can go beyond determination at the species level and can be used to detect more subtle genetic differences such as those leading to a particular resistance profile (see section "Antifungal Susceptibility Testing).
Due to current limitations in sensitivity, and lack of standardized procedures and commercial assays for many rare and emerging fungal pathogens, end-point PCR is generally not included in routine tests of fungal pathogen detection on clinical specimens (Arendrup et al. 2014b). However, efforts are being made to develop new approaches that increase the sensitivity in order to use this powerful technique for direct diagnosis with patient samples. The high sensitivity and specificity that can theoretically be provided by PCR is an incentive, given the generally low amounts of infectious cells present in test specimens. However, several additional limitations may hinder the use of PCR when the DNA template(s) is derived from clinical samples (Schrader et al. 2012). For instance, the presence of hemoglobin and anticoagulants in blood samples that are co-extracted with the DNA inhibit amplification (Zhang, Kermekchiev and Barnes 2010). Certain DNA extraction kits solve this issue by including treatment steps to remove potential inhibitors, which can also be potentially problematic in other approaches. In addition, modified PCR techniques are being developed to overcome limitations, such as low specificity. For example, nested PCR can increase both specificity and sensitivity by making use of two nested primer pairs (Taira et al. 2014).
Analysis of fragment length polymorphisms
Restriction fragment length polymorphism (RFLP) exploits the fact that sequence differences can be identified after digestion with sequence-specific restriction endonucleases. This approach is usually applied in combination with PCR after amplification of the desired DNA fragments. PCR-RFLP has been successfully used to identify different Candida species such as Candida palmioleophila, Candida fermentati, C. albicans, C. dubliniensis, Candida thermophila and C. glabrata (Dendis et al. 2003; Chen et al. 2013; Feng et al. 2014). RFLP analysis requires extensive databases, which impairs its routine use in the clinic. A similar approach, amplified fragment length polymorphism (AFLP), reverses the order of PCR and restriction cleavage (Vos et al. 1995). This technique has been broadly used to evaluate intra-species variation and identify different fungi such as Cryptococcus neoformans/gattii species complex, and Candida species in clinical isolates (Tavanti et al. 2007; Hagen et al. 2015; Calvo et al. 2016; Prakash et al. 2016). Although AFLP is more laborious and expensive than RFLP, it has been shown to be robust, rapid and highly reproducible under standardized experimental conditions (Nyazika et al. 2016). At present, these approaches are predominantly used for the assessment of genetic variation within a species in epidemiological studies.
Real-time PCR
Quantitative PCR (qPCR), formerly designated as real-time PCR, monitors the amount of PCR product by using either fluorescent probes or intercalating dyes (Arya et al. 2005). Dyes (e.g. SYBR Green) are less expensive than probes, but have the disadvantage of binding non-specifically to double-stranded DNA (dsDNA), including primer dimers and non-target DNA (Navarro et al. 2015). To overcome this, melting curve analysis (MCA) can be performed to verify the specificity of a given primer pair. Probes provide intrinsic specificity as they directly bind complementary sequences. Currently, there are several different classes of probes available: primer-probes hairpins (such as Scorpion probes), hybridization probes (e.g. Molecular Beacons), hydrolysis probes (e.g. TaqMan), non-natural bases (Plexor™ primer), as well as probes based on synthetic molecules, such as peptide nucleic acids (PNAs) and locked nucleic acids (LNAs) (Faltin, Zengerle and von Stetten 2013; Navarro et al. 2015). Nowadays, hydrolysis and hybridization probes are extensively used in clinical diagnostics (Wang, Peng and Wang 2014). qPCR-based diagnostic kits for detection of fungal infections are commercially available and provide enhanced specificity and sensitivity compared with traditional identification methods (Table 1). Several kits include identification of the main Candida species. Guidelines such as the minimum information required for the publication of qPCR experiments (Bustin et al. 2009) have facilitated the standardization of these approaches. The main advantage of qPCR over conventional PCR is its ability to provide quantification of the infecting pathogen load, albeit at a higher cost. Although very often a simple positive or negative test for the presence of the pathogen is required in the clinic, knowing the load can be useful in monitoring the effect of the treatment, or when overgrowth rather than simple presence is required to determine infection in a non-sterile human niche. Another diagnostic application of qPCR in the clinic is the monitoring of level of azole resistance in Candida spp. when the main mode of resistance is up-regulation of the gene encoding the target of the drug, or of drug efflux pumps (Sanglard, Ischer and Bille 2001; Torelli et al. 2008; Gohar et al. 2017; Pourakbari et al. 2017), since high levels of transcription of these are associated to azole resistance (Ksiezopolska and Gabaldón 2018).
MCA
MCA aims to discriminate PCR amplicons based on the dissociation kinetics of dsDNA as temperature increases. The melting point (Tm) of dsDNA molecules is defined as the temperature whereby 50% of the molecules have separated into single-stranded DNA. This Tm is sequence-dependent because G-C base pairs form three hydrogen bonds as compared with two in A-T base pairs, and thus the former require more energy to dissociate. Thus, higher Tm is attributed to a higher G/C content. By adding intercalating fluorescent dyes that only exhibit fluorescence when bound to dsDNA, the dissociation process during gradual heating can be recorded as a decrease in fluorescence (Tong and Giffard 2012; Bezdicek et al. 2016). High-resolution melting analysis (HRMA) is considered as the next-generation application of classical MCA (Erdem et al. 2016). HRMA uses brighter dyes at higher concentrations, combined with advanced software and more sophisticated fluorescence-detection systems. HRMA is able to detect and monitor minimal fluorescence variations caused by a change in Tm of less than 0.5°C, which enables detection of sequence differences at a single base pair resolution. The Tm change corresponding to a single G-C replaced by A-T is 41/sequence length °C (Tong and Giffard 2012). Therefore, the length of the amplicon is an important consideration when designing HRMA studies. Short fragments (50–300 bp) will generally result in a well-defined single distinct melting domain and provide straightforward profiles, while longer fragments may depict multiple peaks and reduce the discriminatory power (Wittwer 2003). In addition, the selection of a suitable fluorescent dye is also relevant. Traditional, non-saturating dyes (e.g. SYBR Green) inhibit the polymerase at concentrations that produce maximum fluorescence. A new generation of saturating dyes (e.g. SYTO9, ResoLight) do not have this inhibition effect and therefore can be used at saturation. Another important difference is that non-saturating dyes can re-bind on free sites during the melting procedure, resulting in fuzzier profiles (Duyvejonck et al. 2015). For this reason, saturating dyes are usually preferred over non-saturating ones.
Using MCA of the ITS2 region, Decat et al. succeeded in distinguishing cultured strains of 16 Candida species within 6 h, among them the main pathogenic Candida species (Decat et al. 2013). The commercial multiplex qPCR kit, kiAsperGenius® (Pathonostics B.V., The Netherlands), also makes use of MCA. Besides detecting and differentiating between Aspergillus fumigatus, Aspergillus terreus and Aspergillus spp., this kit provides resistance data about A. fumigatus by detecting resistant-related mutations in the cyp51a gene (White et al. 2015). HRMA has been also used to identify several Candida species (Mandviwala et al. 2010; Arancia et al. 2011; Alnuaimi et al. 2014; Duyvejonck et al. 2015; Nemcova et al. 2015; Bezdicek et al. 2016).
HRMA has been validated by comparison with other techniques such as culture on differential media (Candida ID, CHROMagar), matrix-assisted laser desorption-time of flight mass spectrometry (MALDI-TOF) and nucleic acid sequencing (Alnuaimi et al. 2014; Duyvejonck et al. 2015). Limitations of MCA and HRMA stem from the fact that they use G/C content to discriminate between two different DNA fragments, and therefore do not identify all the sequence variations of the amplicons. For this reason, the pairs of species Candida orthopsilosis and Candida metapsilosis (Decat et al. 2012), and Candida fabianii and Meyerozyma guilliermondii (syn. Candida guilliermondii) (Nemcova et al. 2015) could not be differentiated from each other because of their similar G/C content and overlapping Tm.
The advantages of HRMA technology are a low cost, the use of generic instruments, a low handling time, simplicity and a closed tube format, which minimizes the risk for PCR contamination (Tong and Giffard 2012). Accordingly, HRMA offers a quick and cost-effective method for the simultaneous quantification and identification of the most relevant clinical species of Candida, and for detection of co-infections with these species, directly from clinical samples (Duyvejonck et al. 2015).
Isothermal amplification methods
Isothermal amplification refers to a set of alternative approaches to standard PCR that allows amplification of specific targets without temperature cycles (Qi et al. 2018). Some common advantages of these techniques are their short turnaround time and that they do not require thermocyclers, which make them suitable for clinical settings in low-resource environments (Borst et al. 2002; Zhao and Perlin 2013; Trabasso et al. 2015; Furuie et al. 2016). Isothermal amplification techniques include, among others, nucleic acid sequence-based amplification (NASBA), rolling-circle amplification (RCA), loop-mediated isothermal amplification (LAMP), strand displacement amplification, primer-generation RCA, helicase-dependent amplification, recombinase polymerase amplification, exponential amplification reaction, and whole-genome amplification (Zhao et al. 2015).
Here, we limit our discussion to three of the most promising approaches for fungal diagnostics: RCA, LAMP and NASBA. RCA is a technique based on rolling-circle replication, which occurs naturally for certain types of DNA molecules, e.g. plasmids and viruses with circular genetic material (Novick 1998; Zhao et al. 2015). The resulting product consists of multiple repeats complementary to the template (Ali et al. 2014). This method has already been successfully used for the detection of Candida, Aspergillus and Scedosporium spp. from clinical isolates by targeting species-specific padlock probes to the ITS2 region (Zhou et al. 2008). RCA is fast (2 h) and very specific, but initial set-up costs for probes are high (Wang et al. 2009). LAMP is a DNA amplification technique that facilitates the synthesis of large amounts of DNA using a DNA polymerase with strand-displacement activity that provides high specificity and speed (Mori et al. 2001; Nagamine, Hase and Notomi 2002). LAMP-mediated DNA amplification can be monitored by turbidity since magnesium pyrophosphate is produced proportionally as a result of base incorporation into the newly synthesized DNA strands (Mori et al. 2004). LAMP has already been applied to the detection of pathogenic fungi such as Paracoccidioides brasiliensis (Endo et al. 2004), Pneumocystis spp. (Uemura et al. 2008), C. parapsilosis (Trabasso et al. 2015) and C. albicans (Noguchi et al. 2017). The limit of detection of this technique (1 pg of DNA) (Noguchi et al. 2017) is lower than that of qPCR. However, its lower cost, together with its high specificity and speed, make it a promising tool for the detection of fungal species in clinical samples. Finally, NASBA is a technique that uses continuous amplification of RNA in a single mixture at constant temperature (Compton 1991). It has also been tested for the detection of Candida yeasts (Widjojoatmodjo et al. 1999). An automated system combining NASBA with real-time detection (NucliSENS easyQ® bioMérieux SA) is already in use for bacteria and viruses (Lam et al. 2007; McEwan et al. 2013).
Magnetic resonance and nanoparticles
One of the few currently commercially available methods for diagnosis of Candida infections that does not require culturing is the T2Candida system (T2 Biosystems, USA), which is based on T2 magnetic resonance (Neely et al. 2013). Blood samples are lysed to release DNA, which is then amplified by PCR. Oligonucleotide probes covalently conjugated to superparamagnetic nanoparticles capture the amplified DNA, causing clustering of the nanoparticles. The clustering results in changes to the T2 relaxation time, which is detected by magnetic resonance. This device has achieved 1 colony-forming unit (cfu)/mL sensitivity with time to results of less than 3 h. The T2Candida system can identify C. albicans, C. tropicalis, C. parapsilosis, C. glabrata and P. kudriavzevii. T2Candida is currently undergoing large-scale testing in hospital settings, and several studies have suggested that it is superior to culture and serum methods with regard to sensitivity, specificity and speed (Clancy and Nguyen 2018a). More nanoparticle-based methods are likely to be developed in the future, possibly conjugated to antibodies (Jain et al. 2018).
Large-scale parallel amplification
The broadening spectrum of opportunistic fungal pathogens implies the need to test for the presence of a battery of species, rather than of an individual organism. Combining the parallel amplification of multiple potential targets in a single reaction (multiplexing) has the obvious advantage of saving time, energy and cost, and it is particularly suited for reference laboratories processing huge numbers of clinical samples. Although manual optimization of primers and conditions can successfully result in a combined test for a few potential targets, some specific technologies allow the number of combined targets to be raised to high-throughput scales. Luminex xMAP technology consists of dyed latex microspheres covered with bio-molecular reporters able to hybridize with the targets. A combination of different dyes facilitates 500 simultaneous PCR amplifications from a single sample (Dunbar 2006). Upon completion of the reactions, microspheres are passed through a flowing fluid stream where they are excited by two lasers with wavelengths specific to the internal microsphere dyes and to the reporter molecule attached to the surface. Applications of this technology include single nucleotide polymorphism (SNP) discrimination, screening of genetic diseases, gene expression profiling and microbial diagnostics (Fulton et al. 1997; Colinas, Bellisario and Pass 2000; Spiro, Lowe and Brown 2000; Yang, Tran and Wang 2001; Lee et al. 2004). Successful implementations of this technology for a diverse range of yeast genera including Candida, Cryptococcus and Malassezia have been reported (Diaz et al. 2006; Bovers et al. 2007; Balada-Llasat et al. 2012). Landlinger et al. highlighted the ability of the Luminex system to identify 29 species from 9 fungal genera (Candida,Cryptococcus,Trichosporon,Aspergillus,Mucor,Rhizopus,Penicillium,Absidia and Acremonium) using probes targeting the ITS2 region (Landlinger et al. 2009).
Multiplex ligation-dependent probe amplification (MLPA) is a technique that indicates the presence of multiple DNA target sequences by specific probe amplifications using only a single primer pair (Schouten et al. 2002). Amplification of specific target probes depends on a ligation event that in turn depends on correct hybridization to target DNA. This is achieved by splitting each DNA probe into two oligonucleotides containing either the forward or reverse primer sequences at their ends. These oligonucleotides only ligate to each other after they have fully hybridized to the target DNA sequence, followed by the formation of a PCR product. The same primer pair can be used with multiple probes, allowing the amplification/detection of several targets simultaneously (Dunbar 2006). In multiplexing, specific amplicons are distinguished by size, therefore it is common to adjust the size of the probe forming oligonucleotides with irrelevant sequences (Schouten et al. 2002). MLPA has great potential for species identification and intraspecies differentiation (Pham Thanh et al. 2013).
Droplet digital PCR performs multiple independent PCRs using water-oil emulsion droplets (Vogelstein and Kinzler 1999). A sample is fractionated into thousands (e.g. ∼20 000) of droplets, each containing all necessary PCR reagents. Droplets are passed through a flow cytometer where a positive or negative result is recorded (Roberts et al. 2013). Some of the benefits include multiplexing, speed, cost-effectiveness, high precision and accuracy, as well as the ability to use lower amounts of sample. This platform has been exploited in various fields ranging from detection of ocular infection agents to oncology (Roberts et al. 2013; Chaudhuri et al. 2016; Shembekar et al. 2016). In addition, this approach has been successfully used to identify Aspergillus species in non-clinical contexts (Hua et al. 2018).
DNA hybridization
Hybridization of fluorescent probes to target specific DNA sequences can be applied for molecular diagnostics. Probes can be used directly on clinical specimens and observed by microscopy or in combination with other techniques, such as PCR, and measured with a fluorimeter (DeLong, Wickham and Pace 1989). Fluorescent in situ hybridization (FISH) is based on hybridization of fluorescent probes with taxon-specific regions of the DNA of microorganisms and subsequent detection by fluorescence microscopy or flow cytometry (FC) (Amann, Krumholz and Stahl 1990; Wallner, Amann and Beisker 1993). Probes can be based on natural nucleotides, but an increasing number of approaches exploit the use of nucleic acids analogs. These are artificial nucleic acids chemically modified at the nucleobase, the sugar ring or the phosphodiester backbone. Different forms of modified nucleic acid are available, such as PNA, which contain an uncharged pseudopeptide backbone replacing the sugar phosphate backbone of DNA (Nielsen et al. 1991; Egholm et al. 1993). They can be used in the diagnosis of infectious diseases and detection of resistance-related mutations as they possess unique traits missing from traditional probes such as resistance to nucleases, high temperatures and changes in ionic strength (Chen et al. 2011). PNA probes are commonly used as therapeutic agents that act by altering gene expression but can also be used in the diagnosis of infections. Their suitability for detecting Candida species from blood samples has been tested, showing that they enable identification from blood cultures in about 5 h (Heil et al. 2012). Commercially available kits based on PNA-FISH targeting the rRNA gene are available for the identification of Candida spp., some of which have shown more than 92% sensitivity and specificity with turnaround times of 90 min (Stone et al. 2013; Radic et al. 2016). LNAs are synthetic RNA molecules with a modified ribose ring that increases affinity to complementary DNA or RNA sequences. Consequently, LNA-based probes can provide high specificity and sensitivity. Although they are still not widely used in the clinic, there have been promising developments with respect to their use in detecting diverse fungal species (Ruthig and Deridder 2012; Montone 2014; Ikenaga et al. 2016).
An alternative hybridization-based approach is the hybridization of extracted DNA (or RNA) of a sample to probes fixed on a solid surface (arrays). The advantage of this approach is that the presence of several targets can be interrogated in parallel, allowing the simultaneous identification of a panel of pathogens. Several panels have been developed that focus on invasive or superficial mycoses (Leinberger et al. 2005; Spiess et al. 2007; Sato et al. 2010), and those that combine the detection of both fungal and bacterial pathogens (Cao et al. 2018).
Detection of SNPs
Detecting variants at a single nucleotide can have a high clinical value, particularly if the variant is associated with drug resistance. PCR-based methods can be adapted to detect SNPs with high specificity. These approaches generally rely on one of the following strategies: (i) using specific primers matched to the particular nucleotide variation, or using oligonucleotides to block or clamp the non-targeted template; (ii) using MCA, which is combined with real-time PCR using hydrolysis probes, hybridization probes or dsDNA-binding fluorescent dye; or (iii) design of specific endonuclease digestions of amplified fragments that are able to distinguish the targeted variation. Allele-specific PCR (ASP) makes use of the preferential amplification of desired alleles using Taq DNA polymerase with primers containing allele-specific 3′ ends (Bottema and Sommer 1993). ASP can identify single base changes, as well as small insertions and deletions. Similar techniques include amplification-specific PCR, and the amplification refractory mutation system (ARMS) (Bottema and Sommer 1993). A combination of ASP with quantitative PCR (AS-qRT-PCR) and droplet PCR (AS-Droplet-PCR) may improve the genotyping and quantitation of chimerism in recipients (Taira et al. 2015) compared with regular short tandem repeat PCR. In addition, hybridization with SNP-specific probes can also be used. DNA array systems, which combine parallel hybridization with multiple probes, could provide a fast and easy platform in diagnostic settings. However, a common limitation of all these methods is that they require comprehensive knowledge of key SNPs.
All these techniques have been used to screen for resistance mutations in several fungal pathogens, including array-based systems (De Backer et al. 2001; Garaizar et al. 2006). MCA was used to discriminate C. albicans isolates with and without hotspot mutations in ERG11 that are known to confer azole resistance (Loeffler et al. 2000; Caban et al. 2016). Several PCR-based methods were adapted for SNP detection from clinical samples. For instance, PCR assays were developed to detect echinocandin resistance mutations in the FKS1 and FKS2 genes in C. glabrata (Dudiuk et al. 2014), and in FKS1 in C. albicans (Balashov, Park and Perlin 2006). In addition, mutations in FKS1 and FKS2 in C. glabrata have been identified using MCA (Zhao et al. 2016) and Luminex technology (Pham et al. 2014). HRMA has been used to discriminate among five polymorphic variants in the ERG11 gene of C. albicans (Ge et al. 2010; Caban et al. 2016). Finally, other approaches that are able to detect SNPs have the potential to be used to detect resistance-conferring mutations (Berard et al. 2004).
In summary, molecular approaches that are able to detect resistance directly in clinical specimens require substantial further development. The few assays commercially available lack clinical evaluation. However, resistance screening of clinical specimens is becoming increasingly important as resistance rates are on the rise. Additionally, molecular approaches only provide evidence for the presence of known resistance mutations, but cannot rule out resistance established through unknown mutations or involving other types of biological mechanisms, such as biofilm formation. Therefore, conventional susceptibility testing (see section "Antifungal Susceptibility Testing") will remain an important approach to profiling resistant phenotypes.
SPECTROSCOPY-BASED METHODS
Spectroscopy techniques exploit and detect the interaction of radiation with matter. Currently, different spectrometric approaches are used in microbial diagnostics, including MALDI-TOF MS, Fourrier-transformed infrared spectroscopy (FT-IR), Raman spectroscopy and magnetic resonance spectroscopy (Maquelin et al. 2002; Himmelreich et al. 2003; Wenning and Scherer 2013; De Carolis et al. 2014).
MALDI-TOF MS
MS is a semi-quantitative to quantitative method using ionization of matter and subsequent separation of the ions based on their mass-to-charge ratio. In MALDI-TOF MS analyte molecules are embedded in a (commonly solid) matrix, where they are ionized by a pulsed laser. The resulting ions are separated in the gas phase according to their mass-to-charge ratio, and measured, which results in specific spectra. Over the last two decades, this technique has become a routine tool for the rapid, accurate and sensitive identification of microorganisms in clinical laboratories (Mellmann et al. 2009; Giebel et al. 2010; Seng et al. 2010; De Carolis et al. 2014; Vlek et al. 2014; Kostrzewa 2016). Most of such procedures detect differences in ribosomal proteins, as they are the most abundant in the cells (Suarez et al. 2013). Generally, the use of MALDI-TOF MS for microorganism identification requires culturing and some preparation to embed the biological material into the matrix. Starting from positive blood cultures the whole identification process, including sample preparation and measurement, can be completed in only 1 h (Fraser et al. 2016). To identify microorganisms the profile spectra are compared with a reference database. Such databases are commercially available, e.g. MALDI Biotyper system (Bruker Daltonik GmbH, Germany) (Seng et al. 2010; De Carolis et al. 2014) or Vitek MS (BioMeriéux, Marcy l’Etoile, France). Several studies suggest that MALDI-TOF MS is one of the most accurate and rapid tools for identifying many different yeasts as well as important filamentous fungi (Marklein et al. 2009; Emonet et al. 2010; Santos et al. 2010; Vlek et al. 2014; Taj-Aldeen et al. 2014a). A study comparing the MALDI-TOF MS with standard procedures for 267 clinical isolates including Candida species showed that MALDI-TOF MS correctly identified 92.5% of them (Marklein et al. 2009). Of note, the 20 unidentified species were absent from the reference database, which could be resolved by adding the corresponding spectra. Hitherto, numerous studies have shown the potential of MALDI-TOF MS in identification of different Candida spp. including C. albicans, C. glabrata, P. kudriavzevii, C. parapsilosis, C. tropicalis, C. dubliniensis, C. metapsilosis, C. orthopsilosis,Kluyveromyces marxianus and Candida nivariensis (Qian et al. 2008; Taj-Aldeen et al. 2014a; Angeletti et al. 2015; Galán et al. 2015). Updating the reference database with spectra from different isolates of the target species is relatively easy and could be the fastest solution to diagnose emerging pathogens. This has been demonstrated for C. auris and related species, whereby MALDI-TOF MS has been shown to have a high accuracy in distinguishing between Candida haemulonii and C. auris (Cendejas-Bueno et al. 2012; Kathuria et al. 2015), and even in detecting variation within a single species, allowing epidemiological studies in C. auris (Girard et al. 2016; Prakash et al. 2016). Other studies have shown the potential of MALDI-TOF MS to identify isolates at the species and subspecies levels in the Cr. neoformans/gattii complex (Posteraro et al. 2012; Hagen et al. 2015), for Trichosporon,Geotrichum and related genera (Kolecka et al. 2013), and for Malassezia spp. (Kolecka et al. 2014; Denis et al. 2017). Moreover, using this technique Taj-Aldeen et al. have achieved 100% accuracy for identification of more than 200 clinical isolates, including 11 belonging to uncommon yeast species (Taj-Aldeen et al. 2014b). Overall, these studies illustrate that MALDI-TOF MS is a rapid, accurate and reliable method for identification of microorganisms, including yeasts. The technology is now being developed for detection of antifungal resistance (Kostrzewa and Pranada 2016) (see section "Antifungal Susceptibility Testing").
Other MS methods, FT-IR spectroscopy and Raman spectroscopy
Electrospray ionization MS coupled with broad-spectrum PCR (PCR/ESI-MS) is a promising technique for microbial diagnosis (Jordana-Lluch et al. 2013). In this approach amplicons derived from a PCR are analyzed by MS and the results are matched to a database, which enables the detection of several microbes in parallel. It displays a high sensitivity (lower limit of detection = 1 microbial genome/sample) and thus may be used to detect microorganisms directly from patients’ samples (Jordana-Lluch et al. 2013). Benchmarking performance using blood culture as gold standard gave an overall agreement of 94.2% (Jordana-Lluch et al. 2013). However, the complexity of this technique renders it difficult to integrate into the standard laboratory workflow (Florio 2015).
Rapid evaporative ionization MS (REIMS) has also been successfully applied to identify yeasts (Strittmatter et al. 2013; Bolt et al. 2016). This approach performs mass spectrometric analysis of aerosols originated from heating up cells in the sample, forming gas-phase ions of metabolites and lipids. In contrast to other mass spectrometry methods, REIMS is able to perform the analysis directly from a culture plate, without any extraction or pre-treatment. Published assessments show a 100% accuracy in the identification of 153 clinical Candida isolates, fully agreeing with MALDI-TOF MS and sequencing of the ITS region (Cameron et al. 2016).
Other options for future development of a valid method for rapid and accurate identification of fungal infections are based on vibrational spectroscopy. FT-IR spectroscopy is based on the fingerprint generated by the absorption profile in the infrared light spectrum by microbial cells and can be applied to microbial identification and typing (Quintelas et al. 2018). The technique has been used in some studies to identify Candida species (Timmins et al. 1998; Taha et al. 2013). While the complex information enables typing at even intra-species level of microorganisms (Dinkelacker et al. 2018), it also requires strict standardization of cultivation conditions.
Another method based on vibrational spectroscopy, Raman spectroscopy, is used to investigate the molecular composition of a sample, leading to the formation of spectroscopic fingerprints that can be used to differentiate microbial species and strains (Almarashi et al. 2012; Stöckel et al. 2016). This approach offers several advantages, as it requires minimal biomass and sample handling while enabling a rapid and accurate analysis (Ibelings et al. 2005). The laser irradiates the microbial sample and generates an infinitesimal amount of Raman scattered light, which is detected as a Raman spectrum. A clinical study on intensive care unit (ICU) patients with peritonitis showed a high accuracy (90%) for Raman-based identification of yeasts with only a single misidentification of one C. albicans isolate as the closely related C. dubliniensis (Ibelings et al. 2005). A variation of Raman spectroscopy is surface-enhanced resonance Raman spectroscopy (SERRS) (Faulds, Smith and Graham 2005). This technique consists of PCR-based amplification with multiple DNA probes, each specific for a different fungal pathogen and tagged with specific dyes, followed by exonuclease digestion of dsDNA. This results in the digestion of the bound probes, and the undigested probes are detected by the specific sensors, thereby revealing the pathogens present in the sample. This method reached 100% accuracy when compared with culture-based methods (Yoo et al. 2011).
ANTIBODY-BASED TECHNIQUES
In response to infection, the adaptive immune system of vertebrates can produce tailored defense proteins (antibodies, Ab) that recognize specific molecules of the invading agent (i.e. antigen). Antigen binding may lead to immobilization or neutralization of the antigen and flags it for attack by other components of the immune system. Hence, antibodies provide high specificity and sensitivity that can be exploited for diagnostic purposes. Additionally, there have been efforts to develop therapeutic or protective antibodies for yeast infections (Casadevall and Pirofski 2012; Elluru, Kaveri and Bayry 2015).
Enzyme-linked immunosorbent assay
This approach is based on the detection of low amounts of antigens (down to pico-nanograms/mL) by linking the binding with an antibody with a measurable enzymatic reaction. The technology can be used directly with various clinical samples. Most formats used consist of direct or indirect enzyme-linked immunosorbent assay (ELISA), depending on whether the measurable signal is directly produced by a primary antibody or indirectly by a polyclonal secondary antibody, respectively. Indirect ELISA provides higher sensitivity due to multiple binding of the polyclonal secondary antibody leading to signal amplification. Additionally, sandwich ELISA first captures the antigen by an antibody that is immobilized on a microtiter plate. Capture of the antigen from a complex sample can increase the sensitivity, and might be even necessary if direct coating efficiency of the target antigen is very low (Yolken 1985). The disadvantage of sandwich formats is the need for two antigen-specific antibodies that recognize different areas (epitopes) of the target antigen and that do not compete for antigen binding. In addition to antigen detection, ELISA is used for detection of (invasive) disease-related antibodies in serum samples. In this case, antigen coated to a microtiter plate is used to capture antibodies from human blood serum samples (Clancy et al. 2008).
Currently, sandwich ELISA kits for detection of Candida mannan antigen, a non-covalently attached immune-dominant cell wall protein, in serum samples are commercially available for diagnosis of invasive candidiasis (e.g. Platelia Candida Ag Plus, Bio-Rad). However, evaluations have shown that the Candida mannan ELISA alone has a low sensitivity for predicting invasive candidiasis. The sensitivity of the diagnostic procedure can be significantly increased by additionally testing the sample for presence of host anti-Candida mannan antibodies by ELISA (Mikulska et al. 2010; Held et al. 2013; León et al. 2016). Sensitivity of both ELISAs depends on the species, and was shown to be highest for C. albicans, followed by C. glabrata and C. tropicalis (Mikulska et al. 2010; Held et al. 2013). For detection of the polysaccharide galactomannan as a biomarker of invasive infection with filamentous Aspergillus species, a similar antigen ELISA assay (Platelia Aspergillus Ag, BioRad) is commercially available and recommended for testing serum or bronchoalveolar lavage fluid of high-risk patients. (Ullmann et al. 2018). 1,3-β-d-glucan, a major polysaccharide component of the cell wall of many fungal species, can be detected in serum samples by exploiting an enzyme cascade of horseshoe crab amebocyte extract (not Ab-based) and assays are commercially available (e.g. Fungitell, Associates of Cape Cod) (Onishi et al. 2012). The presence of 1,3-β-d-glucan in serum can indicate invasive disease caused by a fungal species with significant amounts of 1,3-β-d-glucan in their cell walls (e.g. Candida spp.,Pneumocystis spp.,Aspergillus spp. but not Cryptococcus spp. or Mucorales) (Miyazaki et al. 1995a, 1995b). In addition, 1,3-β-d-glucan levels in patients can be elevated, for instance, due to administration of antibiotics or other biopharmaceuticals used during hemodialysis or incorporated into surgical gauze (Otto et al. 2013). Therefore, false-positive test results are common in high-risk groups such as ICU patients (Clancy and Nguyen 2013). Recently, a bispecific monoclonal antibody with specificities for 1,3-β-d-glucan and Candida mannan has been developed, although it lacks validation using clinical samples (Zito et al. 2016). Antigen or antibody detection ELISAs have been developed for other invasive yeast infections such as cryptococcosis (Pfaller 2015), histoplasmosis (Azar and Hage 2017), blastomycosis (McBride, Gauthier and Klein 2017), coccidioidomycosis (Galgiani et al. 2016), paracoccidioidomycosis (Mendes et al. 2017), talaromycosis/penicilliosis (Ning et al. 2018) and sporotrichosis (Bonifaz and Tirado-Sánchez 2017).
Immunofluorescence
In immunofluorescence assays, antibodies conjugated to fluorophores are used to detect antigen at low concentrations (down to pico-nanograms/mL). The fluorescent signal from the conjugates can be visualized with a fluorescence microscope or detected with a fluorimeter. In fungal diagnostics, immunofluorescence is relevant for the detection and identification of invasive pathogens by fluorescence microscopy of fixed tissue samples. A major issue in this area is the lack of highly species-specific antibodies, which would enable an unequivocal pathogen identification (Guarner and Brandt 2011). An indirect immunofluorescence assay intended for the diagnosis of invasive candidiasis by antibodies that are specific for mycelial phase antigens has been developed and commercialized by Vircell (Invasive Candidiasis (CAGTA) IFA IgG) (Strauer 1990). However, the clinical relevance of this assay remains to be evaluated (Clancy and Nguyen 2018b).
Latex agglutination assay
Latex agglutination assays are antibody-based diagnostic methods that can be performed without the use of advanced equipment. In this approach, latex beads coated with specific antibodies are mixed with the test sample. The presence of the antigen leads, within minutes, to cross-linking and visible agglutination of the beads. Testing a dilution series of the sample can provide quantitative information. Conversely, beads can be coated with an antigen to detect antigen-specific antibodies (Molina‐Bolívar and Galisteo‐González 2005). Rapid latex agglutination assays with high sensitivity and specificity have been developed for species identification from colony samples of C. albicans, C. glabrata,P. kudriavzevii andC. dubliniensis, as well as for diagnosis of cryptococcosis directly from serum or cerebrospinal fluid of patients with suspected cryptococcal meningitis (Tanner et al. 1994; Freydiere et al. 1997; Quindos et al. 1997; Marot-Leblond et al. 2006; Wang, Yuan and Zhang 2015).
Lateral flow assays
Lateral flow assays are immunochromatographic antigen detection tests. The liquid test sample is applied through a lateral flow device (LFD), consisting of a series of capillary beds that transport the sample liquid by capillary action. The most common formats are the sandwich and the competitive lateral-flow assays. For antigen detection a liquid test sample is applied to a sample pad, or a dipstick is put into the sample liquid. In the sandwich format the sample liquid flows through capillary action to a conjugate pad that contains the first antigen-specific antibodies conjugated to colored particles, most commonly latex micro- or gold nanoparticles. If the antigen is present in the test sample, immune complexes of antigen and antibody–particle conjugate are formed. Then the liquid passes a detection membrane that has a test-line containing the second antigen-specific antibody. Antibody–antigen conjugates are captured at the test-line leading to visible accumulation of the color, indicating a positive result. Usually a control line is embedded into the detection membrane containing a third antibody specific for the first antibody–particle conjugate (not the antigen) (Bahadır and Sezgintürk 2016).
Ideally, LFDs are rapid, affordable, robust and designed for stand-alone detection of biomarkers in clinical samples, without the need for further equipment or sample pre-treatment, which may qualify them for point-of-care use by untrained personnel, or in resource-limited environments. A lateral flow device that fulfills these criteria has been developed and commercialized for the diagnosis of cryptococcosis (CrAg LFA, IMMY). The sandwich lateral flow assay has high sensitivity (98–100%) and specificity (97–100%) in detection of glucuronoxylomannan of all major serotypes in serum, plasma, cerebrospinal fluid and urine as a biomarker for cryptococcosis (Vidal and Boulware 2015). This LFD has been extensively evaluated and approved and is recommended by the World Health Organization for screening and diagnosis of patients at risk (WHO 2015). Recently, an LFD for detection of human host antibodies against C. albicans enolase as a surrogate marker for invasive candidiasis in serum samples has been described. The LFD assay results were in agreement with indirect ELISA detection of anti-enolase antibodies in clinical samples, but larger scale evaluations are necessary before its clinical value can be determined (He et al. 2016).
The use of antibody-based approaches for the diagnostics of yeast infections is promising, but there is a lack of recent developments in this field. A major drawback of antibody-based diagnostics can be low sensitivity, particularly when compared with PCR-based approaches. The sensitivity of antibody-based methods highly depends on the properties of the antibodies available as well as on the actual presence of surrogate biomarkers in a patient sample, where timing of sampling can be critical. While much progress has been made in targeted generation and selection of antibodies, the identification of suitable biomarkers of active infection (as opposed to past infection or colonization) remains problematic. Relevant biomarkers might be present in very low concentrations, or be unstable, rapidly degraded and/or cleared from the circulation. There is also the risk that antibodies cross-react with irrelevant antigens or that antigenic sites are blocked by formation of immune complexes with human host antibodies.
On the other hand, detection of an appropriate biomarker can yield information about the state of disease while detection of pathogen DNA by PCR is no indication for pathogenic activity of a commensal or presence of viable pathogens (see section "Diagnosis of Infection Type and Stage"). Currently available assays for cryptococcosis diagnostics demonstrate that antibody-based detection methods can be extremely sensitive and specific if an appropriate biomarker is available (Pfaller 2015; Vidal and Boulware 2015; Wang, Yuan and Zhang 2015).
While reviewing the literature it became apparent that the efforts for development of antibody-based diagnostics of invasive yeast infections have been very limited. This might also be due to the fact that clinical evaluation is costly and time consuming. Methods that combine the advantages of biomarker detection by antibodies with the sensitivity of PCR (immuno-PCR) were developed in the early 1990s (Sano, Smith and Cantor 1992), but to our knowledge no applications for fungal diagnostics have been published.
Due to their unique structure, single-domain antibodies (Muyldermans 2013) represent an attractive and largely unexplored area for development of fungal diagnostics. Single-domain antibodies can recognize epitopes that are unavailable for common antibodies and therefore can have extraordinary specificities and affinities and might be able to overcome immune complex formation problems. Antibody-functionalized sensors (e.g. surface acoustic wave sensors) represent attractive platforms that are being developed for application in diagnostics and may provide an incentive to pursue entirely new approaches (Turbé et al. 2017).
ANTIFUNGAL SUSCEPTIBILITY TESTING
The growing prevalence of fungal infections is accompanied by an increased use of antifungal drugs (Pfaller and Diekema 2010; Lockhart et al. 2012). Antimycotic agents in clinical use belong to one of a few drug classes including polyenes (i.e. amphotericin B), azole derivatives (i.e. voriconazole, fluconazole, posaconazole and itraconazole), echinocandins (i.e. anidulafungin, caspofungin and micafungin) and flucytosine (Loeffler and Stevens 2003). Susceptibility towards different drugs varies among pathogenic yeast species and, similar to the situation with antibiotic resistance in bacteria, pathogenic yeasts can acquire resistance due to adaptation to drug exposure (Fairlamb et al. 2016; Ksiezopolska and Gabaldón 2018). The emergence of azole- and echinocandin-resistance in Candida spp. is of great concern, as they complicate therapeutic management (Arendrup and Perlin 2014). Particularly, the emergence of echinocandin-resistance in the intrinsically less azole-susceptible species, such as C. glabrata (Grosset et al. 2016; Mccarty et al. 2016), limits treatment options. Hence, assessment of the levels of susceptibility to different antimycotics of the infecting strain is important to guide antifungal therapy.
Broth-based microdilution and plate cultivation methods
The Clinical and Laboratory Standards Institute (CLSI, www.clsi.org) and European Committee on Antimicrobial Susceptibility Testing (EUCAST, www.eucast.org) have developed reference methods to perform antifungal susceptibility tests based on broth microdilution of yeast cultures (Posteraro and Sanguinetti 2014; Song et al. 2015; European Committee on Antimicrobial Susceptibility Testing 2018; Antifungal Susceptibility Testing Files & Resources 2019). Both CLSI and EUCAST procedures serve to define minimal inhibitory concentration values and clinical breakpoints, which are the values that ideally predict treatment success or failure (European Committee on Antimicrobial Susceptibility Testing 2018; CLSI 2019). Several commercial antifungal susceptibility tests have been developed, with the fully automated Vitek 2 system being one of the most widely used (Posteraro and Sanguinetti 2014).
Agar-based cultivation methods to assess antifungal susceptibility are available. One of the classical agar-based assays is disc diffusion, in which a zone of growth inhibition is produced in a plate culture around a paper disk soaked with the antifungal. The Epsilonmeter test (Etest) is commercially available to assess susceptibility of Candida spp. towards antifungal drugs. These methods are cost effective and easy to set up, but results are not always consistent with EUCAST/CLSI standard approaches (Sewell, Pfaller and Barry 1994; Szekely, Johnson and Warnock 1999; Rex et al. 2001; Dannaoui et al. 2010; Alastruey-Izquierdo et al. 2015).
FC and MALDI-TOF approaches for antifungal susceptibility testing
Over the last two decades, the use of FC approaches for rapid antifungal susceptibility testing has been explored. FC measures single cell fluorescence after exposition to a fluorescent dye, and can be used to measure difference in fluorescence in Candida spp. when exposed to different levels of a given antifungal drug. Although this technique is faster than CLSI and EUCAST reference methods, reducing the time for detection from 24–48 h to 4–9 h, it is expensive, technically complicated and has not yet been validated for clinical use. Nevertheless, it has successfully been used to test susceptibility in species such as C. albicans,C. glabrata,P. kudriavzevii and C. parapsilosis (Chaturvedi, Ramani and Pfaller 2004; Mitchell, Hudspeth and Wright 2005; Pina-Vaz et al. 2005; Vale-Silva and Buchta 2006; Benaducci et al. 2015).
MALDI-TOF MS (see section "Spectroscopy-based Methods") has also been used for antibiotic resistance testing in bacteria (Wolters et al. 2011), and there have been recent attempts to extend this to susceptibility testing in yeasts (Marinach et al. 2009; De Carolis et al. 2012b; Vella et al. 2013; Saracli et al. 2015). This approach is based on the detection of mass spectrometric profile changes depending on the antifungal concentration, and it was first applied to test fluconazole susceptibility in C. albicans (Marinach et al. 2009). Other studies have shown a high concordance between MALDI-TOF MS and the CLSI reference method for measurements of susceptibility to echinocandins, or the presence of resistance-conferring mutations in FKS1 (De Carolis et al. 2012a; Vella et al. 2013). More recently it has been used to detect C. albicans, C. glabrata and C. tropicalis strains resistant to various azoles (Saracli et al. 2015) and as a rapid method for the detection of echinocandin susceptibility in C. albicans and C. glabrata (Vatanshenassan et al. 2018) and C. auris (Vatanshenassan et al. 2019).
Nucleic acid-based methods to detect mutations conferring resistance
Resistance can also be acquired through adaptation to drug exposure by species that are naturally susceptible. This secondary resistance is generally conferred by point mutations in the genes encoding proteins targeted by the drug, or by promoter mutations leading to the overexpression of these proteins or of drug efflux pumps (Ksiezopolska and Gabaldón 2018). Amplification of these regions by PCR (see section 2) followed by Sanger sequencing, as well as targeted or whole genome sequencing (see section 7) can directly decode the sequence of drug-resistance related genes and thus detect the existence of mutations potentially conferring resistance in an isolate of interest, even without previous knowledge of such mutations. In addition, any PCR-based or hybridization-based method able to discern single-point mutations (see section 2.8) can be potentially applied to test the existence of a set of known resistance-conferring mutations (De Backer et al. 2001; Frade, Warnock and Arthington-Skaggs 2004; Kofla and Ruhnke 2007; Gygax et al. 2008; Monteiro et al. 2009; Tsai et al. 2010; Posteraro et al. 2017). Probe-based approaches have a higher sensitivity, and require less DNA and lower DNA quality. Therefore, these techniques are promising for resistance screening of clinical specimens. A drawback of all the probe approaches is that a catalog of known resistance-conferring mutations is only partially available, even for the best-studied species, and therefore a negative result does not ensure the absence of resistance. Direct sequencing approaches can discover novel mutations but generally need a fresh culture, therefore delaying the results. However, emerging sequencing technologies may soon allow rapid, non-targeted sequencing directly from patient samples. Approaches for the direct detection of resistance using such next-generation sequencing (NGS) approaches are discussed in section 7.
DIAGNOSIS OF INFECTION TYPE AND STAGE
The identity of an infectious agent and its potential drug susceptibility are important pieces of information that help to guide therapy (see above). In addition, knowledge about the progression of the infection and the physiological state of the microbe, such as whether it is an invasive stage or forming biofilms, could be very valuable for a more precise assessment of the infection. Finally, many opportunistic yeast pathogens are normal components of the human microbiota or our close environments (Gabaldón, Naranjo-Ortíz and Marcet-Houben 2016). Hence, opportunistic yeast pathogens naturally colonize human tissues and can be detected in many samples that are not primarily sterile (such as vaginal swab, urine or sputum). However, species identification tests cannot discriminate between colonizing and infecting strains, which leads to false-positive results (Khot and Fredricks 2009).
Distinguishing colonization from infection
The ability to detect morphological or physiological features that are specifically expressed during the infection stage is seen as an opportunity to differentially diagnose colonizing versus infecting yeasts in tissues that are not primarily sterile. This approach requires finding suitable molecules (i.e. biomarkers) that are only (or differentially) expressed during the infection stages either by the pathogen or the host (Allert, Brunke and Hube 2016; Decker et al. 2017). Opportunistic yeast pathogens such as Candida spp. display specialized physiological programs that are optimized for commensalism and/or pathogenesis in different niches (Neville, d’Enfert and Bougnoux 2015). The exploration and description of such programs is more advanced for C. albicans, where transcriptomic analysis of yeasts interacting with the host have identified some specific genes such as EFH1, SFU1,HOG1 or EFG1, with critical roles in either commensalism or pathogenesis (Hube 2004; White et al. 2007; Neville, d'Enfert and Bougnoux 2015). On the host side, several studies have been performed focusing on how the immune system discriminates between Candida colonization and infection. In the case of C. albicans, differential recognition of yeast and hyphae could be the key to understanding the immune response (Gow et al. 2011). For example, it has been shown that C. albicans hyphae induce inflammasome activation and the subsequent release of active interleukin-1β in macrophages, which can be used to distinguish between colonization and tissue invasion by C. albicans (Cheng et al. 2011).
Ex vivo staining of whole blood for CCR6+CXCR3– T helper cells provides a mean of rapid identification of patients with chronic mucocutaneous candidiasis due to Th17 deficiency, indicating a potential diagnostic application (Dhalla et al. 2016). On the other hand, in oral epithelial cells, hyphae cause damage and induce a biphasic innate immune mitogen-activated protein kinase response (the ‘danger response’), which was found to be critical for identifying and responding to the switch of commensal microbes to pathogenicity and which represents a sensor of pathogenic C. albicans invasion (Moyes et al. 2010). The fungal moiety responsible for the danger response was recently identified as candidalysin, a cytolytic peptide toxin that directly damages epithelial membranes and activates epithelial immunity (Moyes et al. 2016). However, more studies are needed to investigate the diagnostic potential of these findings.
Detection of biofilm formation
Yeasts can form biofilms, which are defined microbial communities, often adhering to a surface, in which cells are attached to one another and embedded within an extracellular matrix (Hall-Stoodley et al. 2012). This architecture provides protection from the environment, allowing survival in hostile conditions, evasion of host immune mechanisms and reduction of the competitive pressure from other microorganisms (Davey and O’Toole 2000; Silva et al. 2009). Additionally, fungal biofilms can minimize the action of drugs, which can be trapped in the extracellular matrix. In some cases fungal biofilms have been reported to be 1000-fold more resistant to antifungal treatments than planktonic cells (Ramage et al. 2001; Di Bonaventura et al. 2006). Biofilms are also problematic because they can grow on medical devices such as catheters, urethral stents or prosthetic implants, adversely affecting the function of the device (Ramage, Martínez and López-Ribot 2006) and serving as a constant source of microbial cells (Cornely et al. 2012). For all these reasons, biofilms are generally associated with chronic infections that persist despite adequate antimicrobial therapy and host immune defenses (Cornely et al. 2012). Finally, biofilm-based yeasts infections have been shown to increase mortality rate (Tumbarello et al. 2012; Rajendran et al. 2016).
Currently available diagnostic tools to detect biofilms in clinical samples are difficult and time consuming, and have high risk of false negatives, mostly due to the difficulty of obtaining a representative sample (Cornely et al. 2012). Thus, the choice of the samples is important for a correct diagnosis (Lipsky et al. 2012; Percival et al. 2012; Høiby et al. 2015). Biofilms can be detected by direct observation of cell aggregates under the microscope, or through staining of cultures. The most common culture-based methodologies are Congo red agar, tube and tissue culture plate (Mathur et al. 2006). The identification of clinical symptoms typical for fungal infections, the recurrence after antifungal therapies and the medical history of biofilm-predisposing conditions are also important factors to assess the presence of the biofilm (Ramage, Robertson and Williams 2014; Høiby et al. 2015). Assessment of the level of biofilm-forming ability of infection-causing strains can be achieved by means of the tetrazolium salt (XTT) assay, a rapid and highly reproducible method for the formation of fungal biofilms that is easily adaptable for antifungal susceptibility testing (Pierce et al. 2008). Advanced microscopy techniques such as confocal laser microscopy and scanning electron microscopy are the most recommended, but they are generally not available in clinical microbiological laboratories (Malic et al. 2009). Another study performed FISH with a universal Cy3-labeled rRNA probe followed by staining with Calcofluor White for filamentous fungal biofilm detection in water (Gonçalves et al. 2006). A rapid and non-invasive method, using novel high-throughput technologies, would present many advantages over current methods. Similar to the above-mentioned signatures of infection, biofilms could be diagnosed by detecting biofilm-associated factors such as expressed transcripts or enzymes (Winter et al. 2016).
TRENDS IN NGS
Recent advances in nucleic acid sequencing technologies have revolutionized the way in which biological research is performed (van Dijk et al. 2014; Goodwin, McPherson and McCombie 2016). Currently, a variety of NGS methods are available (supplementary Table S1 available online, for additional information) with ongoing developments to increase throughput, read length and accuracy. Current technologies range from ‘sequencing-by-synthesis’, implemented by Illumina, which produces short, accurate reads at high throughput, to nanopore-based sequencing, which produces long reads (up to 1 Mb), but with less throughput and accuracy (Goodwin, McPherson and McCombie 2016; supplementary Table S1 available online). The reduction of sequencing cost and the development of novel approaches has facilitated the implementation of NGS outside the research lab, progressively entering the agronomic, forensic and clinical fields (Berkman et al. 2012; Lecuit and Eloit 2014; Børsting and Morling 2015). Although most of NGS applications in clinical microbiology focus on bacteria and viruses, fungi are equally amenable to NGS. Thus, NGS holds the promise of offering novel diagnostics tools for yeast infections (Fig. 3).
Schematic workflow for the implementation of NGS-based utilities in the clinics. From left to right, the scheme demonstrates the workflow of procedures for NGS applications in clinical set-up, including sample isolation and preparation, NGS based on the addressed question, corresponding bioinformatics data analysis and retrieval of the ultimate results. Green and red braces indicate the potential advantages and limitations of clinical NGS, respectively. WGS—whole-genome sequencing; TAS—targeted amplicon sequencing.
NGS applications in the clinics
The most common clinical applications of NGS are generally based on the human genome, with whole-genome or exome sequencing used in cancer prognosis (Luthra et al. 2015) and diagnosis of Mendelian diseases (Jamuar and Tan 2015). Such applications are increasingly being adopted as a routine procedure in healthcare (Roychowdhury and Chinnaiyan 2014; Association of Public Health Laboratories 2015; Hynes et al. 2017). More recently, NGS is increasingly being used in clinical microbiology laboratories (Deurenberg et al. 2017), as its precision outperforms most of the traditional diagnostic approaches (Turabelidze et al. 2013). For instance, it was recently shown that NGS provides more precise HIV diagnosis with a better discrimination between different genotypes (Shaw et al. 2016). Furthermore, it can provide very high sensitivity for the diagnostics of rare pathogens, as shown with the identification of Leptospira as the causative agent of encephalitis after the failure of 38 different diagnostic tests (Wilson et al. 2014). Apart from diagnostics, NGS can also be applied to epidemiological studies, and it has been successfully used in studies of various outbreaks (Reuter et al. 2013; Sherry et al. 2013). Finally, since NGS can provide whole-genome information, it can be used to determine relevant pathogen features such as antimicrobial resistance and virulence characteristics (Wain and Mavrogiorgou 2013; Biswas et al. 2017). Indeed, a previous study revealed that it is possible to predict resistance to therapy with a specificity higher than 95% in Escherichia coli and Klebsiella pneumoniae using NGS tools (Stoesser et al. 2013). Such advantages are particularly relevant for organisms that are difficult to culture, as is the case for many fungi.
This ability of NGS to detect organisms that otherwise would never be found or studied is promoting a better understanding of the human microbiome (Wang et al. 2015), allowing the study of possible correlations between the microbiome composition and disease states (Ji and Nielsen 2015). However, the detection of an organism by NGS does not imply that this organism is alive, or that it is the causative agent of disease. For example, as discussed in the previous section, the detection of an opportunistic pathogen that can also occur as a commensal is not informative for an infection. Nevertheless, integrating such information with the surrounding microbiome can lead to a probable answer regarding its action. For instance, Bittinger et al. demonstrated that relative abundances of bacteria and fungi in the lung are different during fungal colonization and fungal infection, which can be exploited for clinical diagnosis (Bittinger et al. 2014).
NGS applications for yeast infections
Considering the NGS applicabilities described in the previous sub-section, and that yeast species able to cause diseases in humans are diverse (Huffnagle and Noverr 2013; Taj-Aldeen et al. 2014b; Gabaldón, Naranjo-Ortíz and Marcet-Houben 2016; Pande et al. 2017), the extension of NGS technologies to yeast infections is promising. Indeed, it was recently shown that for C. glabrata, whole-genome sequencing allows the parallel detection of several resistance markers at the same time, being a good alternative to several PCR/DNA sequencing reactions (Biswas et al. 2017). In addition, it is worth noting that some pathogenic yeast clades frequently form hybrids (Pryszcz et al. 2014, 2015; Hagen et al. 2015; Schröder et al. 2016), leading to the origin of new lineages that can be neglected or misidentified with conventional diagnostic tools. For instance, NGS allowed the identification of different parental and hybrid lineages in C. orthopsilosis species (Pryszcz et al. 2014; Schröder et al. 2016), which with conventional tools were always considered the same agent of infection. This is of particular concern, because these hybrid lineages present high genomic plasticity that may allow unpredictable adaptations to new environments or conditions (Mixão and Gabaldón 2018).
Even if fungi represent only a small portion of the microbiome, it is an important part, contributing to the equilibrium between all the communities (Qin et al. 2010; Botschuijver et al. 2017). As reviewed by Zoll et al., changes in the mycobiome in immunocompromised patients can lead to opportunistic fungal infections (Badiee and Hashemizadeh 2014; Zoll et al. 2016). NGS is an important tool to assess the emergent properties of the diverse microbial communities (Nguyen, Viscogliosi and Delhaes 2015; Zoll et al. 2016). Metagenomics approaches have been used for monitoring the presence of pathogens and for pathogen detection (Cuomo 2017); however, the complexity of the procedure and the so far long times for diagnosis are preventing its widespread use (Gu, Miller and Chiu 2019). In addition, when studying the complete microbiome through NGS, it is necessary to take into account that fungi represent only 0.1–1.0% of it (Qin et al. 2010; Huffnagle and Noverr 2013), which may decrease their detectability by NGS (Zoll et al. 2016). Amplicon-based approaches based on PCR-based amplification of specific marker regions (such as ITS region of the rDNA locus discussed in section 2) coupled to NGS-based sequencing of the amplicons can be used to study the fungal component of a complex microbial exosystem (Huseyin et al. 2017). However, this approach generally lacks enough resolution to provide a diagnosis at the species level.
Fungal transcriptomics and RNA sequencing
The potential applications of NGS in the clinics, and particularly in yeast infections, go much further than the identification of genomic characteristics of the pathogen. It also facilitates the study of RNA markers, which may be explored to identify the presence of transcripts that, for example, are only expressed during invasive growth (see section 6). In addition, global analysis of gene expression at the RNA level will greatly contribute to the investigation of interaction between the host and the pathogen, and thus will further our understanding of pathogenesis (Hovhannisyan and Gabaldón 2018). With RNA sequencing (RNA-seq), we can obtain tens of thousands of transcripts from one sample, in an unbiased manner (Van Keuren-Jensen, Keats and Craig 2014). This technology has been used to profile the transcriptome of the host, the pathogen or even both (by means of ‘dual RNA-seq’) during infection (Westermann, Gorski and Vogel 2012). Transcripts originated from the host and the pathogen can be distinguished by aligning to the corresponding reference genomes (De Cremer et al. 2013; Enguita et al. 2016), allowing the determination of RNA markers involved in the infection from both sides. For instance, a recent study showed that the expression of hypha-associated secreted enzymes is greatly increased in the yeast C. albicans during mouse vaginal infections, activating the inflammasome in the host (Bruno et al. 2015). Dual RNA-seq is an approach that enables not only the detection of differentially expressed genes, but also the identification of transcriptional regulatory events such as alternative splicing (Van Keuren-Jensen, Keats and Craig 2014). Furthermore, when performed at different time points, this technology serves to reconstruct the dynamics of the infection and host–pathogen interactions (Tierney et al. 2012; Holcomb et al. 2017). A list of selected RNA-seq datasets generated during fungal infection can be found in the supplementary Table S2 available online.
Limitations of NGS for clinical use
NGS technologies can be of high value for clinical laboratories. However, despite the numerous possible applications for clinical mycology, there are still several limiting factors preventing these approaches from becoming routine laboratory techniques. The cost of sequencing machines is still high, which makes this technology affordable only for large medical institutions. The alternative of outsourcing the sequencing of samples necessarily implies delays and increased logistics. Even with the dramatic decrease of sequencing costs in the last decade, the price of these technologies is still high for routine diagnostics purposes (Weymann et al. 2017). In this regard, recent developments are leading to cheaper and smaller devices, such as the portable MinIon from Oxford Nanopore, a sequencing device that weighs less than 100 g and can directly load sequencing results to a laptop. Applications of these technologies for the rapid and specific detection of plant pathogens have been developed (Chalupowicz et al. 2019), and similar approaches are certainly being developed for human pathogens.
RNA-seq presents an additional limiting factor compared with whole-genome sequencing. Although reproducible and reliable data are obtained with this technology, some biases and errors can be introduced during the experimental procedures and data analysis, making it difficult to compare results from different samples (Van Keuren-Jensen, Keats and Craig 2014). This is an important aspect for clinical applications, and possibly, RNA-seq will not be routinely used in the clinical laboratories until it is solved. Current NGS machines produce massive amounts of data, and the storage of the data also presents cost and data privacy issues. Although cloud-based storage of data is more cost-effective than local storage, the privacy of clinical data might be at risk. Finally, the analysis of high-throughput sequencing data is based on sophisticated and careful bioinformatics processing, requiring specific expertise in NGS data analysis. Therefore, to overcome this challenging issue, automatic pipelines and software for NGS data analysis have to be developed, so they can be easily used by clinical laboratories (see section 8 for details). Another important issue is the availability and completeness of specific reference databases for pathogen identification and characterization. While in the scientific community the de novo approach for organism characterization is frequently used, in clinical laboratories a reference-based strategy is preferred (Loeffelholz and Fofanov 2015). For bacterial pathogens, information in these databases is relatively rich, whereas data for fungal pathogens are scarce, and the absence of a high quality reference information can produce misleading results (discussed in Stavrou et al. (2018)). Even in the presence of a reference genome for a species, problems may arise when the clinical strain has substantial genomic re-arrangements or is a hybrid. Thus, in order to use NGS approaches for fungal pathogen identification, the amount of reference data in these databases has to be significantly increased (see section 8 for details).
NGS technologies can have a broad application in the clinic, ranging from diagnostics to outbreak studies and therapeutic decisions. Moreover, with the advances that NGS is making in the study of the human microbiome, it is probable that the future of diagnostics and therapeutic approaches target the study of the microbiome rather than of a single organism, trying to understand the origin of disequilibrium and applying therapeutics to restore it. Therefore, despite the limitations described here, attempts have been made to implement NGS facilities for clinical usage (Goldberg et al. 2015).
BIOINFORMATICS TOOLS AND DATABASES RELEVANT FOR DIAGNOSTICS OF YEAST PATHOGENS
Many of the mentioned technologies such as proteomics or NGS are high throughput and are able to generate large amounts of data on a single run. Such data has to be managed and processed computationally and there is a continuous need for new and more efficient algorithms that are able to manage, analyze and integrate increasing amounts of disparate data. Conversely, the accumulation of such data entails a high potential for new discoveries by integrating accumulated knowledge with newly obtained data. In the field of diagnostics of yeast pathogens, these interconnected needs have driven important developments in terms of bioinformatic tools and databases. Here we will provide an overview of the most important ones.
Pipelines and workflows in fungal -omics
Advances in NGS and other high-throughput -omics technologies have triggered a bioinformatics revolution and the development of tools to manage and mine immense amounts of biological information. These tools no longer consist of a single piece of software coupled with a knowledge database, but rather consist of a complex workflow referred to as a ‘pipeline’, where different tools are sequentially used, including those consulting existing databases. In the context of host–pathogen interactions, pipelines have been designed to analyze NGS data from various pathogens, including fungi, in both clinical and environmental settings. Genomes and transcriptomes of fungal pathogens, and metagenomes (i.e. collective genetic material present in a microbial community) can be reconstructed from raw sequencing reads by graph-based algorithms that assemble the NGS reads into contigs, scaffolds or chromosomes (Miller, Koren and Sutton 2010). Features, like genes, are then predicted and annotated, which can be used to infer the phenotypic potential of the studied pathogen(s). Once an annotated reference genome is available, it can be used as a reference sequence to map newly obtained sequencing reads from genomes or transcriptomes belonging to strains from the same related species. This process is based on read alignment or mapping algorithms (Li and Homer 2010).
The most common re-sequencing data analysis pipelines are mainly oriented to genotyping and comparative transcriptomic analyses by means of RNA-seq. The former is designed to call and annotate mutations at the genome-wide level or via gene panels and capture systems (e.g. the entire set of known resistance genes or resistome) to elucidate potential markers—mainly SNPs and INDELs (insertions and deletions)—related to the evolution, population dynamics, transmissibility and infectivity of fungal pathogens (Wilkening et al. 2013; Cornish and Guda 2015). In RNA-seq analysis for gene expression profiling, there are three main types of pipelines: mRNA-seq, small RNA-seq and dual RNA-seq. The first two are designed to quantify and compare the expression patterns of the mRNAs (both coding and non-coding) and small RNAs expressed by fungal pathogens under a particular condition. The dual RNA-seq pipeline deals with data obtained by simultaneous sequencing of host and pathogen RNA, and thus focuses on the crosstalk between pathogens and their host at different stages of the infection (Enguita et al. 2016; Wang et al. 2016). Other mapping pipelines in fungal research focus on processing reads sequenced via 18S rRNA/ITS amplicons. These pipelines are widely used to obtain diversity landscapes of fungal communities to investigate any plausible interrelation among the abundance distribution of the species inhabiting the fungal community and the characteristics of the sampled environment (White et al. 2013; Cole et al. 2014). In clinics it is necessary to have a consensus on the best practices for NGS data analysis, and therefore some guidelines were published recently (Roy et al. 2018).
Proteomics and metabolomics are two other -omics fields that require implementation of bioinformatics pipelines for identification, functional analysis and quantification of proteins and metabolites associated to fungal infections. The type of data usually obtained are those coming from spectrometric and spectroscopic techniques (see section 3). Together these two techniques result in complex datasets intended to characterize the proteome and/or metabolome profile of the analyzed sample (Kim, Nandakumar and Marten 2007; Winder, Dunn and Goodacre 2011).
The implementation of bioinformatics pipelines in routine application of fungal -omics is still a challenge for many clinical and medical research centers. -Omics technologies generate huge amounts of data, and most centers lack powerful computational infrastructure for these data analysis and storage. Moreover, management of the bioinformatics pipelines and Unix-based command line environments require specific skills in programming and IT systems. However, there are several initiatives (supplementary Table S3 available online), most of them commercial, that aim to provide solutions for managing bioinformatics workflows based on Graphical User Interfaces (GUIs) allowing non-skilled laboratory staff or researchers to quickly and efficiently analyze -omics data and produce easily interpretable results. On the other hand, with the increasing dependence of clinical mycology on bioinformatics, it is clear that it will be highly necessary to recruit specialized bioinformatics personnel, or to subcontract/outsource bioinformatics services to companies in every clinical or research center aiming to make the most of fungal -omics data.
Databases and knowledge platforms devoted to fungal organisms
Over the last decades, the increasingly frequent recourse to -omics and bioinformatics to generate valuable knowledge from the high-throughput data has resulted in a wide repertory of resources that have improved and updated previously existent reference databases. The most popular databases for NGS data annotation are: the non-redundant (nr) and the Reference Sequence (RefSeq) database of the National Center for Biotechnology Information (NCBI Resource Coordinators 2018), the UniProt Knowledgebase (Schneider et al. 2009) and Ensembl (Kersey et al. 2016). For coding sequences, the gene identifiers or accessions provided by the aforementioned databases are correlated with other vocabularies like the Gene Ontology (GO) vocabulary (The Gene Ontology Consortium 2014) or the system of Enzyme Commission (EC) numbers (Bairoch 2000) (http://enzyme.expasy.org), both facilitating a better understanding of the functional role of the annotated gene.
In addition, the progression of fungal -omics and bioinformatics has also promoted the emergence of a wide variety of databases and repositories specifically dedicated to fungi, including yeast pathogens (supplementary Table S4 available online). For instance, the Saccharomyces Genome Database (SGD) collects community resources about the budding yeast Saccharomyces cerevisiae aiming to integrate curated information and experimental results. The Candida Genome Database (CGD) (Skrzypek et al. 2017) is modeled after SGD but contains genomic resources for C. albicans,C. glabrata,C. parapsilosis,C. dubliniensis and other Candida species. Candida Gene Order Browser (CGOB) (Fitzpatrick et al. 2010; Maguire et al. 2013) and Yeast Gene Order Browser (YGOB) (Byrne and Wolfe 2005) provide manually curated homologous genes datasets in Candida species and other yeast species, respectively. EnsemblFungi is more general and contains many genomes for a wide range of fungal organisms, and is a subset of Ensembl genomes (Kersey et al. 2016). MycoCosm (Grigoriev et al. 2014) integrates fungal genomics data and analytical tools to perform comparative genomics. There are other databases that are specialized in recording/storing information on host–pathogen interactions to identify genes and proteins involved in host–pathogen interaction pathways, which could be used as possible targets for new drug discovery. For example, FungiDB (Stajich et al. 2012; Basenko et al. 2018) includes whole-genome sequences and annotations, experimental and environmental isolate sequence data, comparative genomics, analysis of gene expression, as well as supplemental bioinformatics analyses and a web interface for data mining. The Pathogen-Host Interaction database (PHI-base) (Urban et al. 2015, 2017) is devoted to storing and maintaining effectively the vast and growing number of proven genes that have a role in pathogenicity. PHI-base includes a wide variety of hosts and pathogens, and C. albicans and C. glabrata are among them. PHI-base is highly dependent on domain experts to manually curate new gene entries provided by strong experimental evidence (gene disruption experiments) and literature references. The Host-Pathogen Interaction Database (HPIDB) (Kumar and Nanduri 2010; Ammari et al. 2016) stores and maintains protein–protein interactions from diverse mammalian and plant hosts infected by fungi, bacteria and other pathogens.
In addition, according to the International Code of Nomenclature for algae, fungi and plants (ICN) (Turland et al. 2018), it is a mandatory requirement to register fungal names for valid publications. MycoBank (Robert et al. 2013; Turland et al. 2018) is a database to document mycological nomenclatural novelties (new names and combinations) and its associated data. The MycoBank registration system represents a coordination channel between different databases, such as the Index Fungorum and the Fungal Names (http://www.indexfungorum.org/), and it eases the registration process for the scientific community. StrainInfo (Dawyndt et al. 2005; Verslyppe et al. 2014) is an open platform designed to contain all known information about a particular microorganism at the strain level, giving a unique passport to every strain, with a number that traces back to the same isolate, thus providing a uniform overview for all known equivalent strain numbers. The CBS strain database (http://www.westerdijkinstitute.nl/Collections/) and the American Type Culture Collection (ATCC, https://www.lgcstandards-atcc.org/) are another two examples of such repositories. Databases and the integration of their information will certainly constitute the cornerstone of future automated diagnostic applications (see below). Therefore, it is of capital importance to ensure their correct maintenance and to minimize the amount of introduced errors with the use of both manual curation and automated detection of potential errors (see Stavrou et al. (2018) as an example).
Data integration and computationally assisted diagnostics: the last frontier
In the emerging field of personalized and precision medicine, bioinformatics and integrative biology have gained increased attention for their promise to develop more efficient tools for integration and analysis of multi-omics data of complex diseases, including fungal infections and associated clinical data. Integrative analysis of multi-omics data is motivated by the basic idea that to fully understand any biological system, the underlying molecular mechanisms should be considered in the analysis (Kristensen et al. 2014; Rotroff and Motsinger-Reif 2016). Fungal infections are characterized by an interaction between the fungal pathogen and host cells. The integration and analysis of genomic, proteomic and metabolomic data, as well as other meta-data from the patient (i.e. medical record) represents an excellent opportunity to model infection, to make clinically relevant predictions, and to discover biomarkers and target-specific drugs (Durmuş et al. 2015; Larsen et al. 2015; Culibrk, Croft and Tebbutt 2016). Among all the different methodologies that could be used for personalized and precise diagnosis based on multi-omics, the most promising are Bayesian networks (BNs), decision trees, artificial neural networks, nature-inspired and evolutionary algorithms (Larrañaga et al. 2013; Bersanelli et al. 2016). One of the key features of BNs is their ability to propagate probabilities through the network while incorporating uncertainty in the inference process. Of course, a domain expert (e.g. biologist or medical doctor) should be involved in the knowledge discovery process to interpret and validate the final result (Holzinger 2014, 2016; Holzinger and Jurisica 2014; Obermeyer and Emanuel 2016). Some existing tools are BNOmics (Gogoshin, Boerwinkle and Rodin 2017), a reconstruction and modeling framework able to reverse engineer and model networks, and PARADIGM (Vaske et al. 2010), which applies BNs to identify patient-specific pathway activities by means of probabilistic inference (Larrañaga et al. 2013).
Overall, bioinformatics data analysis and knowledge database development are quickly evolving fields in fungal research. Given the continuously dropping costs of NGS and other -omics methods (see section 7), data accumulation, storage, further analysis and interpretation will evidently become more widespread in routine mycology labs. Moreover, the integration of multi-omics data holds a great potential towards understanding host–fungus interactions, potentially allowing the translation of this knowledge into clinical practice, including diagnosis and prognosis. However, today there are still several important challenges, both technical and fundamental, that will need to be overcome.
FUTURE PROSPECTS AND CONCLUDING REMARKS
Ultimately, in the clinical context, the final goal of any of the above mentioned methodologies is its translation into ‘ready-to-use’ systems—also known as point-of-care testing—that can be routinely used at the bedside, without the requirement of sending samples away, and ready to be used and interpreted by clinicians. In such systems the clinical specimen can be directly inserted into a cartridge that contains all the needed chemicals, and the user receives a fully automated report on the identified pathogens. The development of such point-of-care testing has become a trend in diagnostics, including fungal infections. Although developments are still in their infancy, we have here surveyed the most promising devices that can already be found on the market (included in Table 1). The majority of currently commercially available molecular tests are developed to detect the pathogens in whole blood, positive blood cultures, sterile body fluids and tissue samples. Ideally, future developments should include the integration of resistance markers into molecular routine tests, to support physicians in their therapeutic decision.
Although still lagging behind the diagnosis of cancer or viral and bacterial infections, the field of diagnosis of fungal infections is starting to harness recent developments in areas such as proteomics or high-throughput generation sequencing. However, the need for expensive devices and specific expertise, or high running costs are still challenges that need to be overcome for many of the recently developed approaches. Nevertheless, the main players and technology developers have realized the huge potential of the clinical sector and they strive to develop smaller, simpler and cheaper devices. In this regard, the entry into the market of palm-sized sequencing machines and an increasing variety of point-of-care testing devices anticipate a technological revolution in diagnostic devices commercially available for clinical practitioners. Fast and accurate recognition of the infecting agent, even if present in very low amounts, remains the main objective of diagnostics of yeast infections. However, it is important to realize the potential of high-throughput, -omics technologies to go beyond species identification, and provide other relevant information such as potential resistance to antifungals, and the recognition of the disease mode or stage. It would be a huge step forward if we could identify molecular biomarkers specific for colonization or infection and integrate them into novel diagnostic tools that also identify species and potential for resistance. It is unlikely that we will be able to identify a single factor/gene that would satisfy the requirement of such a biomarker, and rather a panel of different genes/factors, potentially from both, pathogen and host, would be required to create such a specific signature. Ideally, such a signature should not only be specific for discriminating between commensalism and infection, but also for the type of infection, infected tissue or fungal species causing the infection. Continuous monitoring of the presence of such biomarkers could follow the progress of the infection and allow a fast reaction to a potential failure of therapy. For this to happen, monitoring should be possible with cheap bedside devices, which could offer both versatility (i.e. serve to detect various biomarkers for species identification, resistance and progress of infection) and specificity. One could imagine that device directly connecting in real time to a central database, whereby gathered information is confronted with previously collected data, including the clinical record of the patient as well as genomic, proteomic and epidemiological information. Algorithms based on artificial intelligence, continuously improved with the help of the doctors’ feedback, will quickly process the data and will provide the doctor not only the readout of the measurements, but also a possible diagnosis and suggested optimal treatment regime, as well as relevant links to other information. These suggestions may seem futuristic but current technologies exist that could perform each of these steps. We need to fill in the gap that exists between sophisticated technologies used at the frontier of research and the needs at the bedside of patients. We need to understand better the biology of infecting microbes, and the process of infection itself, to find suitable biomarkers. We consider that the coming years will witness important advances in these respects.
COLLABORATORS
Members of the OPATHY consortium (in alphabetical order) are: A. Arastehfar1,2, T. Boekhout1,2, G. Butler3, G. Buda De Cesare4, E. Dolk5, T. Gabaldón6,7,8, A. Hafez7,9,10, B. Hube11, F. Hagen1, H. Hovhannisyan6,7, E. Iracane3, M. Kostrzewa12, M. Lackner13, C. Lass-Flörl13, C. Llorens9, V. Mixão6,7, C. Munro4, J. Oliveira-Pacheco3, M. Pekmezovic11, A. Pérez-Hansen13, A. Rodriguez Sanchez14, F.M. Sauer2,5, K. Sparbier12, A.A. Stavrou1,2, M. Vaneechoutte14 and M. Vatanshenassan2,12.
And their affiliations:
1: Westerdijk Fungal Biodiversity Institute, 3584 CT, Utrecht, The Netherlands; 2: Institute for Biodiversity and Ecosystem Dynamics (IBED), University of Amsterdam, 1012 WX, Amsterdam, The Netherlands; 3: School of Biomedical and Biomolecular Science and UCD Conway Institute of Biomolecular and Biomedical Research, Conway Institute, University College Dublin, Belfield, Dublin, Ireland; 4: MRC Centre for Medical Mycology at University of Aberdeen, Institute of Medical Sciences, Foresterhill, Aberdeen, UK; 5: QVQ Holding BV, Yalelaan 1, 3584 CL Utrecht, The Netherlands; 6: Bioinformatics and Genomics Programme, Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Dr. Aiguader 88, 08003 Barcelona, Spain; 7: Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; 8: Institució Catalana de Recerca i Estudis Avançats (ICREA), Passeig Lluís Companys 23, 08010 Barcelona, Spain; 9: Biotechvana, Calle/Catedrático Agustín Escardino No. 9, Scientific Park Universitat de València, 46980 Paterna, Valencia, Spain; 10: Faculty of Computers and Information, Menia University, Egypt;11: Department of Microbial Pathogenicity Mechanisms, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoell Institute (HKI), Jena, Germany; Friedrich Schiller University, Jena, Germany; 12: Bruker Daltonik GmbH, Fahrenheitstr. 4, 28359 Bremen, Germany; 13: Division of Hygiene and Medical Microbiology, Medical University of Innsbruck, Schöpfstrasse 41, 6020 Innsbruck, Austria; 14: Laboratory Bacteriology Research, Department Clinical Chemistry, Microbiology & Immunology, Faculty of Medicine & Health Sciences, Ghent University, Flanders, Belgium; Medical Research Building II, 1st Floor, Ghent University Hospital, Entrance 38, Heymanslaan 10, 9000 Gent, Belgium.
FUNDING
This work was supported by European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. H2020-MSCA-ITN-2014–642 095.
Conflicts of interest. None declared.