 Quality control of 40S ribosome head assembly ensures scanning competence
Quality control of 40S ribosome head assembly ensures scanning competence
The Journal of Cell Biology


H. Ghalei’s present address is Department of Biochemistry, Emory University School of Medicine, Atlanta, GA.

- Altmetric
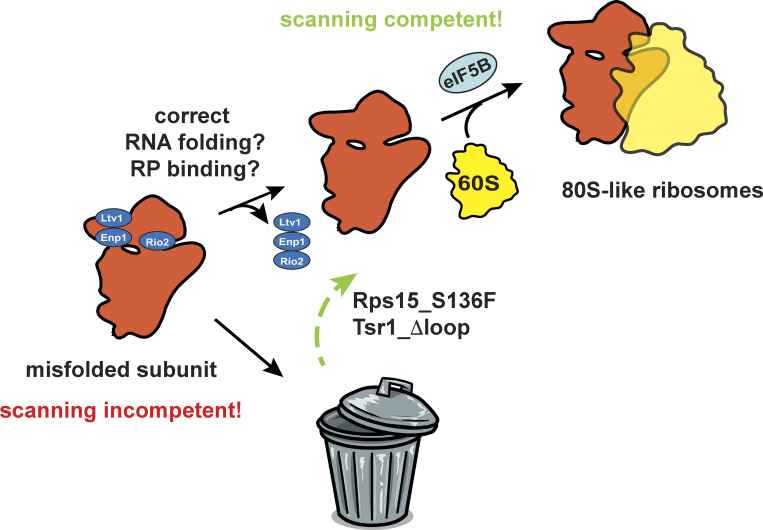
Huang et al. show that formation of 80S-like ribosomes during small subunit maturation is a quality control step that tests head assembly to ensure the fidelity of start-codon selection.
During translation initiation, 40S ribosomes scan the mRNA until they encounter the start codon, where conformational changes produce a translation-competent 80S complex. Destabilizing the scanning complex results in misinitiation at non-AUG codons, demonstrating its importance for fidelity. Here, we use a combination of biochemical and genetic analyses to demonstrate that the ability of the nascent subunit to adopt the scanning complex is tested during assembly via structural mimicry. Specifically, formation of the 80S-like assembly intermediate, which structurally resembles scanning complexes, requires the correct folding of two rRNA elements in the subunit head and the proper positioning of the universally conserved head proteins Rps3, Rps15, Rps20, and Rps29. rRNA misfolding impairs the formation of 80S-like ribosomes, and bypass of individual checkpoints using cancer-associated mutations produces ribosomes defective in accurate start-site selection. Thus, the formation of 80S-like assembly intermediates is a quality control step that ensures scanning competence of the nascent subunit.

Introduction
The 40S ribosomal subunit is a complex structure built of 33 ribosomal proteins (RPs) and one RNA. Previous work in reconstituted bacterial systems demonstrated that folding of the substructures proceeds at different timescales and identified the 40S subunit head as prone to misfolding (Adilakshmi et al., 2008; Duss et al., 2019). Perhaps not coincidentally, during translation, the head is mobile relative to the rest of the subunit (Munro et al., 2009). Thus, the rRNA sequence must encode not just one but multiple head structures, whose stability is regulated by bound ligands such as translation factors and tRNAs. One such conformational change occurs during translation initiation, where mRNA scanning requires an open structure of the head relative to the body (reviewed in Hinnebusch, 2017). Upon recognition of the start codon, the 40S structure closes around the initiator tRNA/mRNA match, ultimately promoting the binding of eIF5B and formation of the translation-competent complex (Hussain et al., 2014; Llácer et al., 2015). Mutations in translation initiation factors and rRNA that stabilize the open structure promote fidelity, while those that stabilize the closed structure lead to misinitiation at near-cognate start codons (e.g., Dong et al., 2017; Fekete et al., 2007; Martin-Marcos et al., 2017; Obayashi et al., 2017; Saini et al., 2010). Thus, the ability to adopt the scanning-competent structure is critical to prevent misinitiation.
40S biogenesis is a multistep process that begins with the cotranscriptional assembly of the majority of the RPs, coupled to the modification and processing of the rRNA. The resulting intermediate is exported into the cytoplasm, where the final rRNA maturation steps are coupled to quality control in a translation-like cycle (Lebaron et al., 2012; Strunk et al., 2012). This cycle involves the joining of mature 60S subunits to premature 40S (pre40S) subunits, mediated by the translation initiation factor eIF5B, leading to the formation of 80S-like ribosomes. These are hubs for quality control, and ribosome maturation (García-Gómez et al., 2014; Ghalei et al., 2017; Lebaron et al., 2012; Strunk et al., 2012). Intriguingly, the structure of 18S rRNA in 80S-like ribosomes resembles the structure of the scanning-competent preinitiation complex (Fig. 1 A). Thus, we hypothesized that the eIF5B-dependent formation of 80S-like ribosomes might be a quality control step to ensure nascent 40S subunits are scanning competent and able to identify start codons with high fidelity.
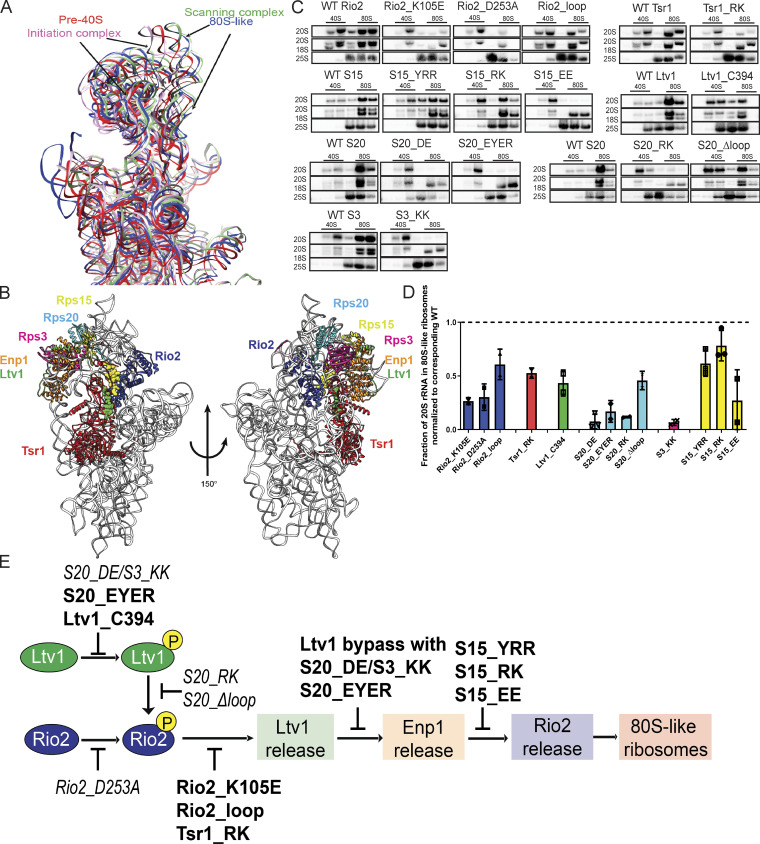
Interface remodeling during the formation of 80S-like ribosomes. (A) Superimposition of 18S rRNA from pre40S (red, PDB accession no. 6FAI; Scaiola et al., 2018), or 80S-like (blue, PDB accession no. 6WDR; Rai et al., 2020 Preprint) ribosome assembly intermediates, and the open and closed conformations of translation initiation complexes (green and pink, PDB accession nos. 3JAQ and 3JAP, respectively; Hussain et al., 2014; Llácer et al., 2015). (B) Composite structure of yeast pre40S subunits (PDB accession no. 6FAI; Scaiola et al., 2018) and human Ltv1 (PDB accession no. 6G18; Ameismeier et al., 2018), obtained by overlaying 18S rRNAs. Only the segment of Ltv1 conserved in yeast is shown in the sphere highlight. The interfaces between the color-highlighted proteins all change in the transition from 40S to 80S-like ribosomes (see Fig. S1 A). (C) Northern blots of 10–50% sucrose gradients from Fap7-depleted cells expressing only plasmid-encoded WT or mutant Rio2, Tsr1, Ltv1, Rps15 (S15), Rps20 (S20), or Rps3 (S3). In all cases Fap7 was depleted in glucose for >16 h. The positions where 40S and 80S ribosomes sediment are indicated. (D) Quantification of the data in C. For comparisons between different yeast strains, mutant proteins were normalized to their corresponding WT protein, as indicated by the dotted line. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (E) Order and dependence of formation of 80S-like ribosomes on Ltv1, Enp1, and Rio2 release. The step impaired by each mutation is shown. Previously described mutations are in italic (not bold) text. Table S4 contains a detailed summary of the yeast strains and plasmids used in each figure.
Here, we use a combination of yeast genetics, biochemical, and structural analyses to demonstrate that the formation of 80S-like ribosomes requires the prior release of the assembly factors (AFs) Ltv1, Enp1, and Rio2. More importantly, these are hierarchically coupled processes that require correct positioning of the universally conserved Rps3, Rps15, Rps20, and Rps29, as well as correct folding of rRNA in the head. Thus, release of these AFs is quality testing the error-prone assembly of the head. Bypass of a specific step in this cycle is promoted by cancer-associated mutations in Rps15, as well as a variant of the AF Tsr1. Ribosomes from cells expressing these variants in Rps15 and Tsr1, as well as other bypass mutants, all exhibit defects in start-codon recognition, but not other steps in translation. Together, these results demonstrate how these AFs set up a checkpoint to prevent ribosomes with misfolded head structures from forming 80S-like ribosomes and thereby being matured. Thus, subunit joining is a checkpoint for scanning competence of small ribosomal subunits.
Results
Point mutations in Ltv1, Rio2, Tsr1, Rps3, Rps20, and Rps15 affect subunit joining
40S ribosome maturation involves a test drive for the nascent molecule to quality control the nascent subunit (Lebaron et al., 2012; Strunk et al., 2012). In this translation-like cycle, mature 60S subunits join pre40S in an eIF5B-dependent manner to form 80S-like ribosomes. These 80S-like ribosomes are required for quality control and maturation, before they are broken apart by the translation termination factor Rli1. Because the formation of 80S-like ribosomes requires a translation initiation factor (Lebaron et al., 2012; Strunk et al., 2012) and is regulated by the kinase Hrr25 (Ghalei et al., 2015; Mitterer et al., 2016; Schäfer et al., 2006), we hypothesized that it is a checkpoint to test translation initiation competence.
Formation of 80S-like ribosomes requires the dissociation of the AF Ltv1 (Ghalei et al., 2015), its interaction partner Enp1, and the kinase Rio2, as well as repositioning of the AFs Tsr1 and Dim1 on the subunit interface (Ghalei et al., 2017; Strunk et al., 2012; Rai et al., 2020 Preprint). These AFs make extensive interactions with each other, as well as the RPs Rps3 (uS3), Rps15 (uS19), and Rps20 (uS10; Fig. 1 B), which are broken in 80S-like ribosomes (Fig. S1 A). Thus, we used a collection of novel and previously identified (Collins et al., 2018; Ferreira-Cerca et al., 2012; Mitterer et al., 2016, 2019) point mutants in these protein–protein interfaces to test if and how they block the formation of 80S-like ribosomes.
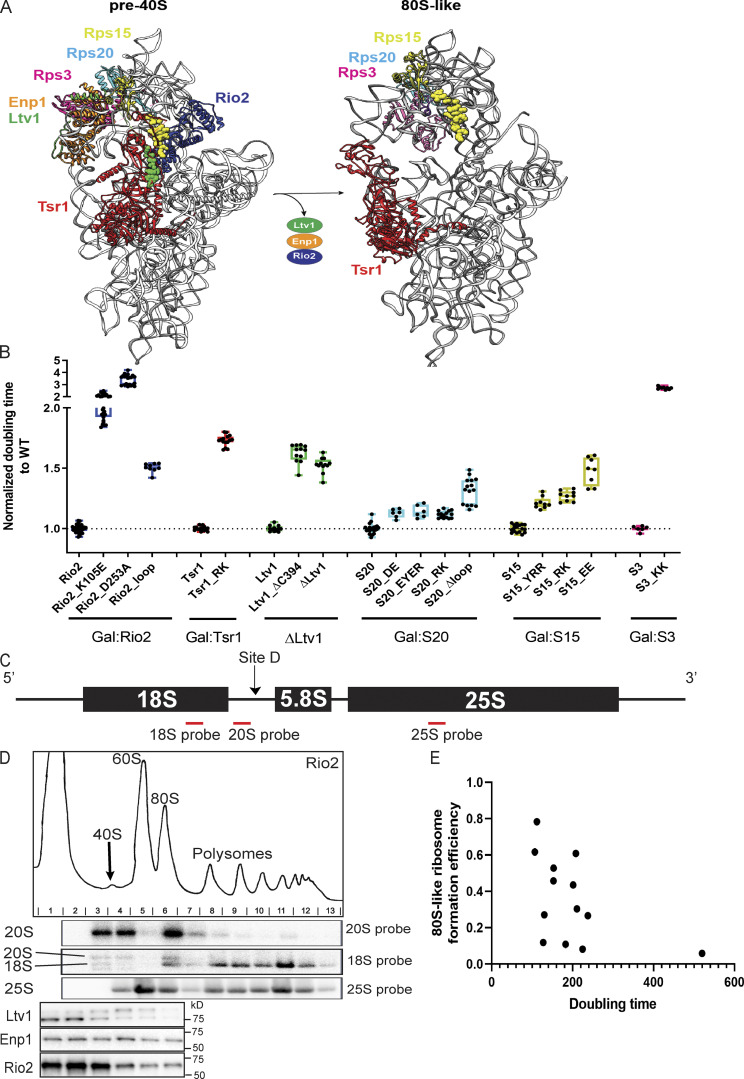
Comparison of protein–protein interfaces in pre40S and 80S-like ribosomes (related to Fig. 1). (A) The figure was constructed from PDB accession nos. 6FAI and 6G18 (left) and 6WDR (right). (B) Normalized doubling time of cells with the indicated WT or mutant proteins. Details of yeast backgrounds and plasmids are described in Table S4. Growth data were collected from at least three biological replicates and two independent measurements. Each mutant passed an unpaired t test with P < 0.0001. (C) Location of Northern probes used in this work. (D) Polysome profiling example of 10–50% sucrose gradients followed by Northern blots and Western blots. Please note that the Western blots shown are the same as those shown in Fig. 3 B bottom section column 3. In this figure, the entire experiment, including polysome profile, Northern blot, and Western blot are shown. (E) The correlation between doubling times and the ability to form 80S-like ribosomes is shown. The correlation coefficient is −0.5.
The mutants increase the doubling time of isogenic yeast strains with the WT gene 1.2- to 3.5-fold (Fig. S1 B). To test if the growth defects were reflective of defects in the formation of 80S-like ribosomes, we used an in vivo assay, where yeast lysates are fractionated over sucrose gradients to separate 40S precursors that accumulate as pre40S or 80S-like intermediates (Fig. 1 C). Gradient fractions were analyzed by Northern blotting for 40S precursors (20S rRNA) and mature subunits (18S and 25S rRNAs; see Fig. S1, C and D) to determine the fraction of 40S precursors that form 80S-like ribosomes. This assay relies on the accumulation of 80S-like ribosomes in yeast depleted of the ATPase Fap7 (Ghalei et al., 2015; Strunk et al., 2012). As we had previously shown that formation of 80S-like ribosomes requires not only eIF5B but also Ltv1 dissociation (Ghalei et al., 2015; Strunk et al., 2012), we reasoned that it could reveal other molecular requirements for formation of 80S-like ribosomes as well. By quantifying the amount of 80S-like ribosomes in the mutant strains relative to their isogenic WT controls (represented by the dashed line in Fig. 1 D, we established that the mutants impair the formation of 80S-like ribosomes (Fig. 1, C and D). Furthermore, we note that the magnitude of the effect on the formation of 80S-like ribosomes correlates with the magnitude of the growth defect from each mutant (Fig. S1 E).
Hierarchical release of Ltv1, Enp1, and Rio2 from pre40S subunits
To investigate the order in which Ltv1, Enp1, and Rio2 are released from pre40S subunits before the formation of 80S-like intermediates, we used the same sucrose-gradient analysis described above and assessed the dissociation of AFs using Western blotting (Fig. S1 D). In Fap7-depleted but otherwise WT cells, Ltv1, Enp1, and Rio2 are largely in the free fraction, as the 80S-like intermediate that accumulates does not have these factors bound (Fig. 2 B; Ghalei et al., 2015; Strunk et al., 2012). To determine if any mutant blocks assembly before or after release of Ltv1, Enp1, and Rio2, we quantified the amount of free Ltv1, Enp1, and Rio2 in the mutant relative to the isogenic WT control. All gradients were replicated with at least two biological replicates to ensure consistency and reproducibility. Furthermore, in each case, we compared a corresponding Fap7-depleted but otherwise WT strain grown in parallel to the Fap7-depleted mutant strain.
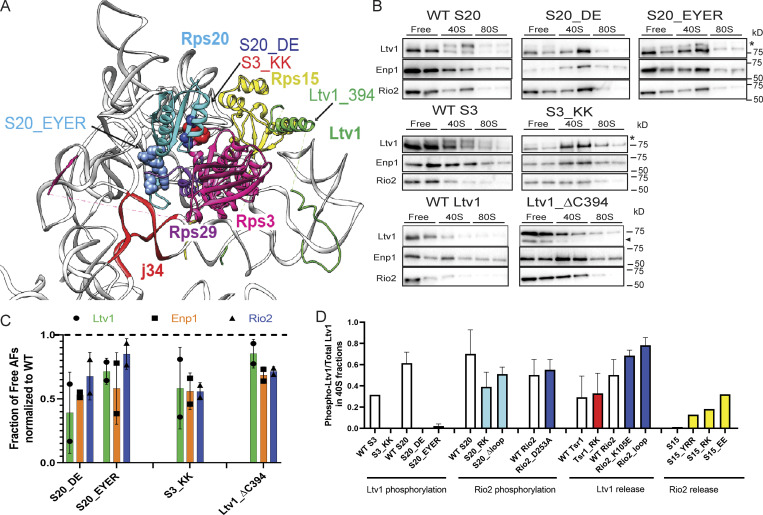
Ltv1 phosphorylation is sensitive to mutations in Rps3, Rps20, and Rps29. (A) Detail from the structure in Fig. 1 B. Mutations in Rps20 and Rps3 are highlighted in sphere. The site of the C-terminal truncation in Ltv1 is indicated. J34: three-way junction between h34-35-38). (B) Western blots for Ltv1, Enp1 and Rio2 of 10–50% sucrose gradients from Fap7 depleted cells expressing only plasmid-encoded WT or mutant Rps20 (S20), Rps3 (S3), or Ltv1. The position where free, 40S-bound, or 80S-bound proteins sediment is indicated. The asterisk marks phosphorylated Ltv1, and the arrowhead points to a degradation product. (C) Quantification of the data in B. To allow for comparison between different yeast strains, each mutant was normalized to the corresponding WT protein, as indicated by the dotted line. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (D) Quantification of phosphorylated Ltv1 in the pre40S fraction.
As described in detail in the next several sections, we identified four classes of mutants: (1) those where Ltv1, Enp1, or Rio2 are not released (Fig. 2); (2) those where AFs are not released but Ltv1 is phosphorylated (Fig. 3); (3) those where Ltv1 is released normally but Enp1 and Rio2 are not (Fig. 4); and (4) those where Ltv1 and Enp1 are released but Rio2 remains bound to pre40S (Fig. 5). Together, these data provide strong evidence for an ordered release of Ltv1, Enp1, and Rio2, with Ltv1 phosphorylation preceding its release.
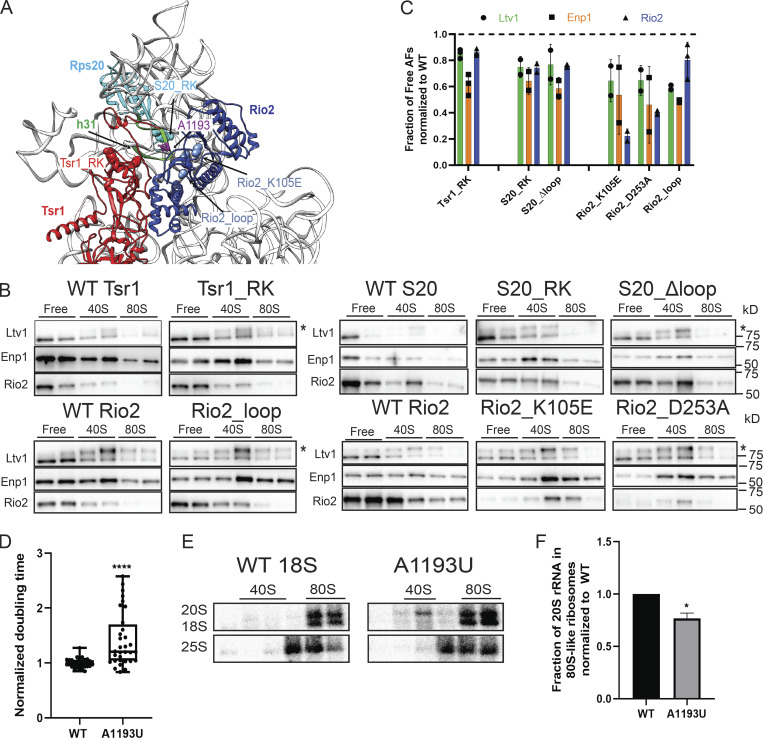
Ltv1 release depends on the structure around helix 31. (A) A composite structure of yeast pre40S (PDB accession no. 6FAI) and mature 40S (PDB accession no. 3JAQ) overlaid on Rps18. Mutations in Rps20, Rio2, and Tsr1 are highlighted in sphere. The loop containing the residues mutated in Rio2_loop is unresolved in all structures, and the flanking residues, R129 and S145, are highlighted in sphere. Helix31 (h31) from mature 40S, which is not resolved in pre40S, is highlighted in green. A1193 is highlighted in purple ball and stick. (B) Western blots for Ltv1, Enp1, and Rio2 of 10–50% sucrose gradients from Fap7 depleted cells expressing only plasmid-encoded WT or mutant Tsr1, S20, or Rio2. The asterisk marks phosphorylated Ltv1. Please note that the Western blots shown in column 3 of the bottom section are also shown in Fig. S1 D, which also displays the polysome gradient profile and the Northern blots for this experiment. (C) Quantification of the data in B. To allow for comparison between different yeast strains, each mutant was normalized to the corresponding WT protein, indicated with the dotted line. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (D) Normalized doubling time of cells expressing only plasmid-encoded WT or A1193U 18S rRNA. Significance was tested using an unpaired t test. n = 32, ****, P < 0.0001. (E) Northern blots of 10–50% sucrose gradients of cell lysates from Gal:Fap7 cells supplemented with plasmids encoding WT or A1193U 18S rRNA (in the background of genomically encoded rRNA). Cells were grown in glucose for over 16 h. 20S and 18S rRNAs were visualized using a probe for a unique tag in plasmid-encoded 18S rRNA (Jeeninga et al., 1997). (F) Quantification of the data in E. Each experiment has two biological replicates. Data are shown as mean with standard deviation. Significance was tested using an unpaired t test. *, P < 0.05.
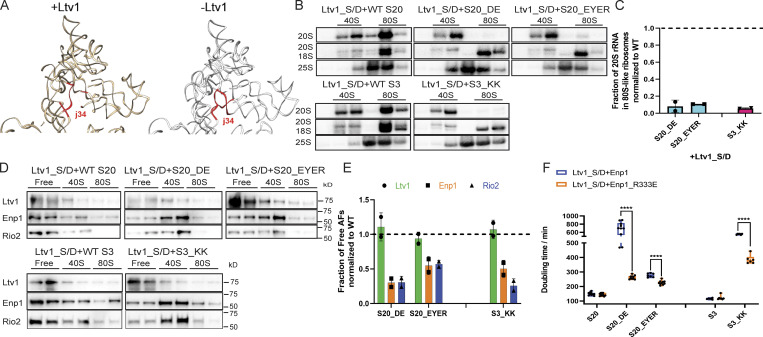
Ltv1 release leads to j34 folding. (A) Comparison of rRNA structures in Ltv1-containing earlier (PDB accession no. 6EML; Heuer et al., 2017) and Ltv1-depleted later (PDB accession no. 6FAI; Scaiola et al., 2018) 40S assembly intermediates. The three-way junction between h34–h35–h38 (j34) is highlighted in red. (B) Northern blots of 10–50% sucrose gradients from Fap7-depleted cells expressing only plasmid-encoded Ltv1_S/D and WT or mutant S20 or S3. In all cases Fap7 was depleted in glucose for over 16 h. (C) Quantification of the data in C. For comparisons between different yeast strains, mutant proteins were normalized to their corresponding WT protein. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (D) Western blots for Ltv1, Enp1 and Rio2 of gradients in B. (E) Quantification of the data in D. For comparisons between different yeast strains, mutant proteins were normalized to their corresponding WT protein. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (F) Doubling time of ΔLtv1,Gal:Enp1,Gal:S20 or ΔLtv1,Gal:Enp1,Gal:S3 cells supplemented with Ltv1_S/D and Enp1 or Enp1_R333E and WT or mutant S20 and S3, respectively. Significance was tested using a two-way ANOVA test. n ≥ 6; ****, P < 0.0001.
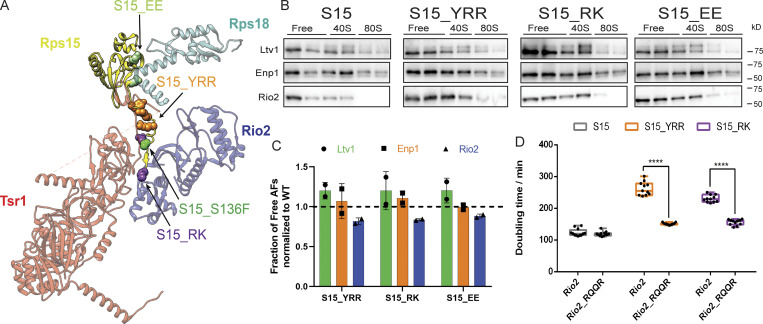
Rps15 mutations block Rio2 release. (A) Detail from the structure in Fig. 1 B. Mutated residues are highlighted in sphere. (B) Western blots for Ltv1, Enp1 and Rio2 of 10–50% sucrose gradients from Fap7 depleted cells expressing only plasmid-encoded WT or mutant Rps15 (S15). In all cases, Fap7 was depleted in glucose for >16 h. (C) Quantification of the data in B. To allow for comparison between different yeast strains, each mutant was normalized to the corresponding WT protein, as indicated by the dotted line. Each experiment has at least two biological replicates. Data are shown as mean with standard deviation. (D) Doubling time of Gal:Rio2,Gal:S15 cells supplied with WT Rio2 or Rio2_RQQR plasmids and WT or mutant S15. Significance was tested using a two-way ANOVA test. n ≥ 9; ****, P < 0.0001.
To gain insight into the sequence of events before Ltv1 release, we considered two previous observations. First, the Rio2_D253A mutant, which blocks autophosphorylation by mutation of the phosphorylation site, blocks not only Rio2 release but also Ltv1 release (Ferreira-Cerca et al., 2014; Ferreira-Cerca et al., 2012; Mitterer et al., 2016, 2019). In addition, mutating the Ltv1 phospho-sites to alanines blocks not only Ltv1 but also Rio2 release (Ghalei et al., 2015). Thus, Rio2 and Ltv1 phosphorylation are both required for Ltv1 release. Additional data described below indicate that Ltv1 phosphorylation (but not release) is required for Rio2 phosphorylation (see Cancer-associated Rps15 mutations below). Together, these data can be summarized in the mechanistic scheme shown in Fig. 1 E.
Fig. 1 E details the extensive exchange across the nascent 40S subunit required for the phosphorylation and release of Ltv1, Enp1, and Rio2 and the formation of 80S-like ribosomes. We next set out to describe in full the data that went into the model in Fig. 1 E and decipher which parts of the nascent subunit were required for each of these steps.
Phosphorylation of Ltv1 is sensitive to mutations in Rps3, Rps15, Rps20, and Rps29
To be recognized by its kinase Hrr25, Ltv1 must be properly positioned on the pre40S. Ltv1 binds directly to Enp1, Rps3, Rps20, and Rps15 (Campbell and Karbstein, 2011; Ghalei et al., 2015; Mitterer et al., 2016), and given its location on ribosomes relative to these proteins, it appears likely that Rps29 also binds Ltv1. This is consistent with synthetic growth defects from Ltv1 deletion and Rps29 C-terminal tagging (Collins et al., 2018). Thus, we wondered if Ltv1 phosphorylation might be sensitive to the binding and positioning of these RPs.
Phosphorylation leads to a significant upshift in the Ltv1 band on an SDS-PAGE gel (Ghalei et al., 2015; Mitterer et al., 2019; Schäfer et al., 2006). Thus, Ltv1 that accumulates on pre40S intermediates in the phosphorylated form will display an upshifted band, while unphosphorylated Ltv1 runs at its normal electrophoretic mobility (Mitterer et al., 2019). Analyzing the collection of mutants, we observed that phosphorylation of Ltv1 is impaired only by Rps3_KK, Rps20_DE, Rps20_EYER, and Ltv1_ΔC394 (truncation of Ltv1 at residue 394, abolishing its binding to Rps15; Fig. 2). These mutants are in the interface between Rps3 and Rps20 (Rps3_KK, Rps20_DE) and Rps20 and Rps29 (Rps20_EYER; Fig. 2 A) and affect proper positioning of Rps3, and likely Rps20, as well as the recruitment of Rps29 (Collins et al., 2018) and positioning of Rps15 (Ltv1_ΔC394). Thus, Ltv1 phosphorylation requires its interactions with Rps3, Rps20, Rps29, and Rps15, thereby probing the correct positioning of Rps3, Rps20, Rps29, and Rps15.
Consistent with these results, it was previously shown that recruitment of Hrr25 is impaired in the Rps3_KK and Rps20_DE mutants (Mitterer et al., 2019). Because Rps29 is located under Rps20 and Rps3 and thus less accessible to Hrr25, and because Ltv1_ΔC394 also blocks the same step, we suggest that Hrr25 recruitment is sensitive to the positioning of Ltv1, which in turn probes the positioning of Rps3, Rps15, Rps20, and Rps29.
Release of Ltv1 is sensitive to mutations around h31
We next investigated our collection of mutants for those where phosphorylated Ltv1 was accumulated on pre40S subunits. These would then be defective in the communication between Rio2 and Ltv1 either before or after Rio2 phosphorylation (Fig. 1 E) to effect the release of phosphorylated Ltv1. As expected from mutations that block the communication between Rio2 and Ltv1, this class includes the Rio2 mutants Rio2_K105E and Rio2_loop (Fig. 3, A–C), the Rps20 mutants Rps20_RK and Rps20_Δloop as well as Tsr1_RK (Fig. 3, A–C). Defective release of phosphorylated Ltv1 in Rps20_RK and Rps20_Δloop has been independently confirmed (Mitterer et al., 2019).
These mutants are all clustered around the tip of helix 31 (h31). This region remains unfolded in pre40S ribosomes (Heuer et al., 2017; Scaiola et al., 2018) but is folded in 80S-like ribosomes (Fig. 3 A; Rai et al., 2020 Preprint). Thus, these mutations may perturb the location or folding of h31, indicating that release of Ltv1 probes the positioning or folding of h31. To test this hypothesis, we created a point mutation in the tip of h31, A1193U. The impact of this mutation on cellular growth was tested in a yeast strain where the transcription of endogenous rRNA can be turned off using a temperature-sensitive mutation in RNA polymerase I (Nogi et al., 1993), and rRNA is expressed from a PGK1-promoter–driven plasmid. Quantitative growth assays demonstrate that this mutation causes an ∼1.5-fold growth defect, similar to previous observations (Fig. 3 D; Nemoto et al., 2010). Previous work also demonstrated accumulation of 20S prerRNA, the precursor to 18S rRNA (Nemoto et al., 2010), indicating that the mutation affects 40S maturation. To test if it impairs subunit joining, we performed sucrose-gradient analysis in a Fap7-depleted strain but without blocking transcription of endogenous rRNA. Sedimentation of the plasmid-derived rRNA was followed by probing against the unique tag contained in the plasmid copy (Jeeninga et al., 1997). Control experiments indicate that gradients from yeast lacking the plasmid do not give a signal, indicating the specificity of the Northern probe (data not shown). This analysis shows that the A1193U mutation in h31 of 18S rRNA indeed impairs subunit joining (Fig. 3, E and F). Thus, perturbations of h31 via the Tsr1_RK, Rio2_K105E, Rio2_loop, Rps20_Δloop, and Rps20_RK mutants, which surround h31, or the A1193U mutation in h31 all impair the release of Ltv1 and thereby the formation of 80S-like ribosomes.
Enp1 release requires folding of h34
The structures of pre40S ribosomes provide insight into the communication between Ltv1 and Enp1 release. These include an atomic model of pre40S subunits purified via a TAP-tag on Ltv1, thereby ensuring full occupancy of Ltv1 (Heuer et al., 2017). In addition, a very similar structure obtained from later pre40S intermediates purified using a tagged version of inactive Nob1 appears to have only partial Ltv1 occupancy (Scaiola et al., 2018). The major difference between these structures is the folding of the junction between h34, h35, and h38 (j34), which is unfolded in the Ltv1-bound sample and folded in the largely Ltv1-free sample (Fig. 4 A). We therefore hypothesized that Ltv1 dissociation leads to folding of this three-way junction, thereby promoting Enp1 dissociation via the associated rearrangements in the pre40S head.
To test this model, we took advantage of the observation that the folded junction is stabilized by Rps3, Rps20 and Rps29 (Fig. 2 A). Thus, weakening their interactions and binding will destabilize j34 folding, and if j34 folding is a requirement for Enp1 release, block Enp1 dissociation. Rps20_DE and Rps3_KK mutate the Rps20/Rps3 interface, while Rps20_EYER affects the Rps20/Rps29 interface. As described above, these mutants block Ltv1 phosphorylation, thereby stalling the assembly cascade upstream of Enp1 release. To bypass this step and isolate potential downstream effects on regulated AF release, we used the phosphomimetic Ltv1 mutant, Ltv1_S/D. This mutant bypasses the requirement for Hrr25, the kinase that phosphorylates Ltv1 (Ghalei et al., 2015).
Consistent with assembly defects in addition to Ltv1 phosphorylation, Rps3_KK, Rps20_DE and Rps20_EYER retain some growth defects in the background of the phosphomimetic Ltv1_S/D (Fig. S2 A). To assess whether they block subunit joining, we combined these mutations with Fap7 depletion and performed polysome/Northern analysis as described above. These data demonstrate that formation of 80S-like ribosomes is impaired in the Rps3_KK, Rps20_DE, and Rps20_EYER mutants, even if the requirement for Ltv1 phosphorylation is bypassed with the Ltv1_S/D mutation (Fig. 4, B and C). Finally, Western analysis demonstrates that the Rps3_KK, Rps20_DE, and Rps20_EYER mutants block Enp1 release (and the downstream Rio2 release), but not Ltv1 release (Fig. 4, D and E).
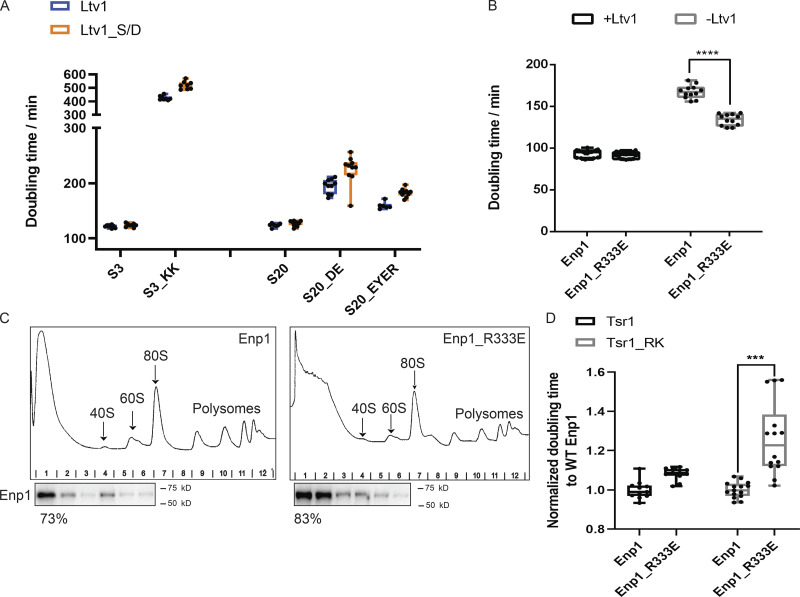
Bypass of Ltv1 phosphorylation defects with the Ltv1_S/D mutation isolates defects on Enp1 release (related to Fig. 4). (A) Doubling times of ΔLtv1,Gal:S3 or ΔLtv1,Gal:S20 cells supplemented with WT Ltv1 or Ltv1_S/D and S3/S3_KK or S20/S20_DE/S20_EYER plasmids, respectively. n ≥ 6. (B) Doubling times of ΔLtv1,Gal:Enp1 cells supplemented with WT Ltv1 or empty vector and WT Enp1 or Enp1_R333E plasmids. Significance was tested using an unpaired test. n ≥ 12; ****, P < 0.0001. (C) Western blots for Enp1 of 10–50% sucrose gradients from cells supplied with WT Enp1 or Enp1_R333E plasmids. The fractions of Enp1 that is unbound and sediments on top of the gradient is indicated below the corresponding blots. This experiment has been repeated twice. (D) Normalized doubling time of Rio2-TAP,Gal:Tsr1,Gal:Enp1 cells supplied with WT Tsr1 or Tsr1_RK and WT Enp1 or Enp1_R333E plasmid. Significance was tested using a two-way ANOVA test. n ≥ 11; ***, P < 0.001.
To further test if these mutations affect Enp1 release, we asked if they would be rescued by a weakly binding variant of Enp1. To design such mutants in Enp1, we identified residues at the Enp1/18S rRNA interface. These were then screened in an assay that takes advantage of the observation that overexpression of Enp1 under the promoter of the yeast TEF2 gene is deleterious in the absence of Ltv1 (Collins et al., 2018). Notably, this observation alone supports the model that Enp1 release is blocked in ΔLtv1 cells. This identified Enp1_R333E as a mutant that has essentially no growth defect on its own, but rescues the defect from Ltv1 deletion (Fig. S2 B). Sucrose-gradient analysis in the presence of Fap7 shows that the mutation increases the amount of Enp1 in the unbound fraction (Fig. S2 C), thus confirming its weak binding.
We next compared the growth of the Rps20_DE, Rps20_EYER, and Rps3_KK mutants (combined with Ltv1_S/D) in the presence of WT and R333E Enp1. As predicted if these mutations block Enp1 release, their growth defects are rescued in the presence of Enp1_R333E, while this mutation has no effect on the growth of WT Rps20 or Rps3. In addition, Enp1_R333E exacerbates the growth defects from Tsr1_RK (Figs. 4 F and S2 D), demonstrating the specificity of its rescue.
Thus, this combination of biochemical and genetic data demonstrates that the interactions among Rps3, Rps20, and Rps29, which stabilize the folding of the h34–h35–h38 junction, are required for Enp1 release, supporting the structure-based model that Ltv1 release leads to j34 folding, which is required for Enp1 release.
Mutations in Rps15 affect Rio2 release
Quantification of Western blots from the sucrose gradients shows that in the final class of mutants Ltv1 and Enp1 were released from the pre40S intermediate as in the isogenic WT strains, while Rio2 was depleted from the free fraction as it remained bound to pre40S ribosomes. Thus, this class of mutants, which includes mutations in the C-terminal tail of Rps15, Rps15_YRR, and Rps15_RK, as well mutations in the Rps15/Rps18 interface, Rps15_EE (Fig. 5, A–C), appears to specifically block the release of Rio2.
To confirm that these mutations directly affect Rio2 release, we tested whether their effects on cell growth could be rescued by self-releasing Rio2 mutants. Indeed, gradient centrifugation in the presence of Fap7 indicates that that relative to WT Rio2, Rio2_RQQR (R17Q20Q24R27E) is partially redistributed to the free form (Fig. S3 A), as expected from a weakly binding mutant. We next tested whether Rio2_RQQR could rescue the growth defects from the Rps15 mutations. Comparison of the doubling times of Rps15_YRR and Rps15_RK in the presence of WT Rio2 or Rio2_RQQR demonstrates that Rio2_RQQR rescues the growth defects of these mutations, while it has no effect in a WT background (Fig. 5 D). This effect is specific to these Rps15 mutants and not observed with other tested mutations, with the exception of Rio2_D253A, Rps20_RK, and Rps20_loop (Fig. S4, B–E). These mutants all block Rio2 phosphorylation (Ferreira-Cerca et al., 2012; Mitterer et al., 2019; see below).
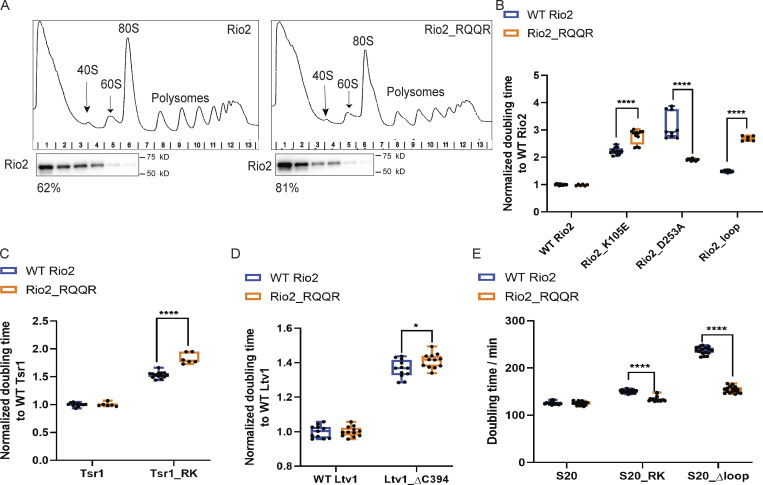
Identification of a weakly binding Rio2 mutant (related to Fig. 5). (A) Western blots for Rio2 of 10–50% sucrose gradients from cells expressing only plasmid-encoded WT Rio2 or Rio2_RQQR. The fraction of Rio2 that is unbound and on top of the gradient is indicated below the corresponding blots. This experiment was repeated twice. (B) Normalized doubling times of Gal:Rio2 cells supplied with WT Rio2 or the indicated Rio2 mutants. Significance was tested using a two-way ANOVA test. n ≥ 6; ****, P < 0.0001. (C) Normalized doubling times of Gal:Rio2,Gal:Tsr1 cells supplied with WT Rio2 or Rio2_RQQR and WT Tsr1 or Tsr1_RK plasmids. Significance was tested using a two-way ANOVA test. n ≥ 6; ****, P < 0.0001. (D) Normalized doubling times of ΔLtv1,Gal:Rio2 cells supplied with WT Rio2 or Rio2_RQQR and WT Ltv1 or Ltv1_ΔC394 plasmids. Significance was tested using a two-way ANOVA test. n ≥ 11; *, P < 0.05. (E) Doubling times of Gal:Rio2,Gal:S20 cells supplied with WT Rio2 or Rio2_RQQR and WT S20 or S20_RK or S20_Δloop plasmids. Significance was tested using a two-way ANOVA test. n ≥ 12; ****, P < 0.0001.
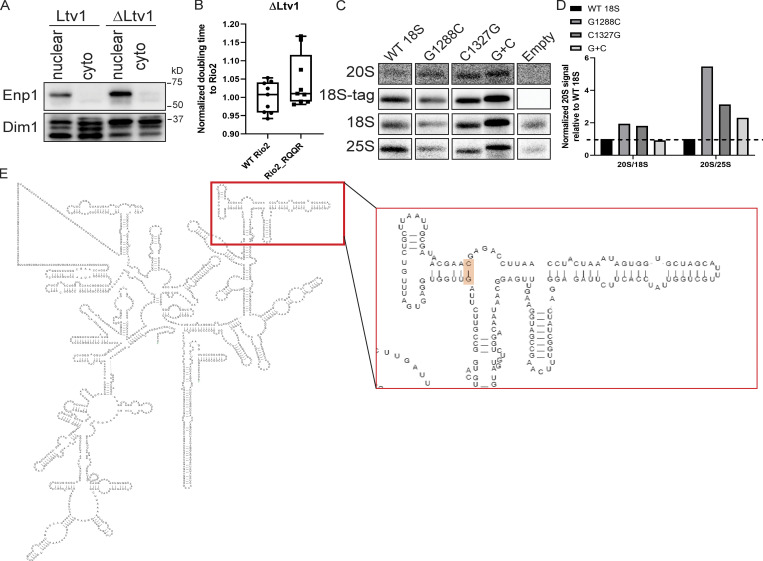
Probing Enp1 release and j34 folding (related to Fig. 6). (A) Western blots for Enp1 and Dim1 of the nuclear and cytoplasmic fractions in Fig. 6, B and C. (B) Normalized doubling times of ΔLtv1,Gal:Rio2 cells supplied with WT Rio2 or Rio2_RQQR plasmids. Significance was tested using an unpaired t test. n = 9. (C) Northern blots of total cell lysates of NOY504 cells supplied with plasmids encoding WT 18S or the indicated 18S rRNA mutants, grown at 37°C. (D) Quantification of Northern blots in C. Secondary structure of 18S rRNA is shown, highlighting the location of the G1288/C1327 base pair.
These data strongly suggest that correct positioning of Rps15 is required for communicating Enp1 release to Rio2 to promote its dissociation, consistent with Rio2 binding Rps15 directly (Campbell and Karbstein, 2011). Existing structures or biochemical data do not show direct interactions between Enp1 and Rps15 (Campbell and Karbstein, 2011; Heuer et al., 2017; Scaiola et al., 2018), indicating that Enp1 release may affect Rps15 via the RNA structure.
rRNA misfolding blocks entry into the translation-like cycle
Taken together, the data above show that formation of 80S-like ribosomes requires proper positioning of RPs and folding of head rRNA, suggesting that entry into the translation-like cycle monitors the structural integrity of the nascent 40S head (Fig. 6 A). To further test this conclusion, we wanted to assess the consequences from misfolding of these key elements. Dimethylsulfate footprinting indicates that j34 is misfolded when ribosomes are assembled in the absence of Ltv1 (Collins et al., 2018). If release of AFs and the formation of 80S-like ribosomes monitors misfolding of j34, then we predict that j34 misfolding blocks Enp1 release.
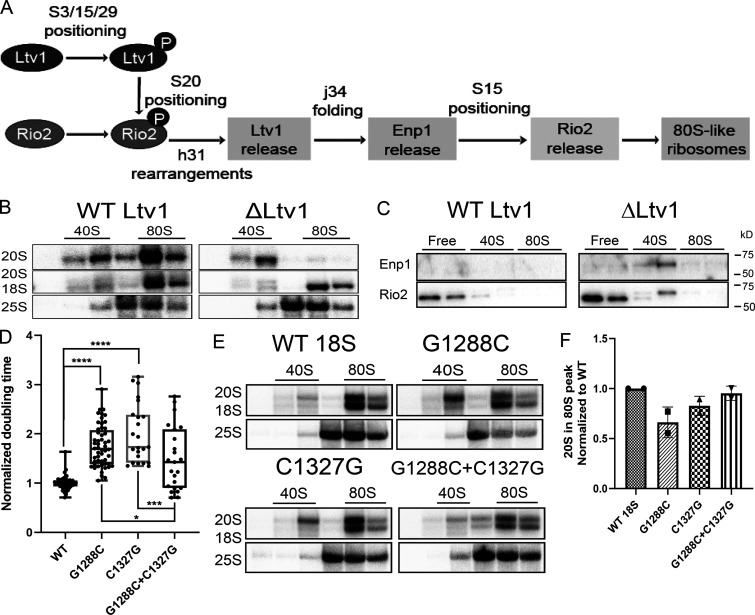
rRNA misfolding impairs the formation of 80S-like ribosomes. (A) Order and dependence of formation of 80S-like ribosomes on Ltv1, Enp1, and Rio2 release. The assembly step checked at each point is indicated. (B) Northern blots of 10–50% sucrose gradients from the cytoplasmic fraction of ΔLtv1,Gal:Fap7 cells supplemented with a plasmid encoding WT Ltv1 or an empty vector and Fap7 depleted in glucose for >16 h. (C) Western for blots for Enp1 and Rio2 of gradients in Fig. 6 B. Note that the doublet for Rio2 arises from proteolysis during the subcellular fractionation. (D) Normalized doubling time of cells expressing only plasmid-encoded WT or mutant 18S rRNA. Significance was tested using a one-way ANOVA test. n ≥ 23; *, P < 0.05; ***; P < 0.001; ****, P < 0.0001. (E) Northern blots of 10–50% sucrose gradients of cell lysates from Gal:Fap7 cells supplemented with plasmids encoding WT or mutant 18S rRNA (in the background of genomically encoded rRNA). Cells were grown in glucose for >16 h. 20S and 18S rRNAs were visualized using a probe for a tag in plasmid-encoded 18S rRNA (Jeeninga et al., 1997). (F) Quantification of the data in E. Each mutant was normalized to the corresponding WT protein. Each experiment has two biological replicates. Data are shown as mean with standard deviation.
To test this prediction, we first tested if Ltv1 deletion blocks subunit joining. Ltv1 promotes nuclear export of nascent pre40S (Seiser et al., 2006). Because nuclear intermediates cannot form 80S-like ribosomes, we used nuclear/cytoplasmic fractionation to isolate the cytoplasmic fraction (Fig. S4 A). Sucrose-gradient analysis and Northern blotting of the cytoplasmic portion demonstrate that in ΔLtv1 cells grown in the absence of Fap7, subunit joining is impaired (Fig. 6 B), as expected if misfolding blocks release of Enp1. Gradient analysis demonstrates that Rio2 is accumulated on pre40S subunits in the ΔLtv1 cells (Fig. 6 C). Unfortunately, during cellular fractionation, Enp1 dissociates and degrades from the cytoplasmic intermediate (Fig. S4 A). While this precludes a quantification of its release defect, we note persistence of Enp1 in pre40S in the ΔLtv1 cells, consistent with a block of Enp1 release (Fig. 6 C). Furthermore, the weakly binding Enp1_R333E (Fig. S2 B), but not the weakly binding Rio2_RQQR (Fig. S4 B), rescues the growth defect from Ltv1 deletion, supporting the model that Ltv1 deletion directly blocks Enp1 release and Rio2 release only indirectly.
To further test if rRNA misfolding blocks the formation of 80S-like ribosomes, we created point mutations in the closing base pair of h35, directly adjacent to j34 (Fig. S4 E). We used the same plasmid-encoded system described above to test if these mutations affect yeast growth. Mutations of one nucleotide on each side of the helix provides ∼1.5–2-fold growth defect, which is largely rescued when the two mutations are combined to restore base pairing (Fig. 6 D). Northern blotting demonstrates that the effect arises from defects in 40S maturation (Fig. S4, C and D). Finally, individual mutations in the closing base pair adjacent to j34 reduce the formation of 80S-like ribosomes, which is restored in the compensatory mutant (Fig. 6, E and F).
Together, these data demonstrate that correct folding of the h34–h35–h38 junction is required for Enp1 release and the formation of 80S-like ribosomes, strongly supporting the notion that the formation of 80S-like ribosomes is part of a quality control mechanism to ensure only correctly assembled 40S subunits are matured. To test if this was part of a quality control mechanism that ensures the integrity of the ribosome pool, we looked for suppressors that would allow for bypass of one of the assembly blocks that we identified in here.
Cancer-associated Rps15 mutations and Tsr1-mutations allow for bypass of h31 defects
Mutations in the C-terminal tail of Rps15 occur in ~20% of all patients suffering from chronic lymphocytic leukemia (CLL) before treatment (Bretones et al., 2018; Ljungström et al., 2016). These mutations are associated with an aggressive form of the disease and cluster in the same region of Rps15 associated with defects in Rio2 release (Fig. 5). Because the CLL-associated mutations appear to promote cancer, we hypothesized that they might promote bypass of individual steps in the AF release cascade.
To test this hypothesis, we created the analogous point mutations in yeast by making Rps15_T134A, Rps15_T135F, and Rps15_S136F, as well as the triple mutant Rps15_TTS (Fig. S5 A). These mutants have no discernible effect on cell growth (Fig. S5 B) or the efficiency of 80S-like ribosome formation (Fig. S5, C and D).
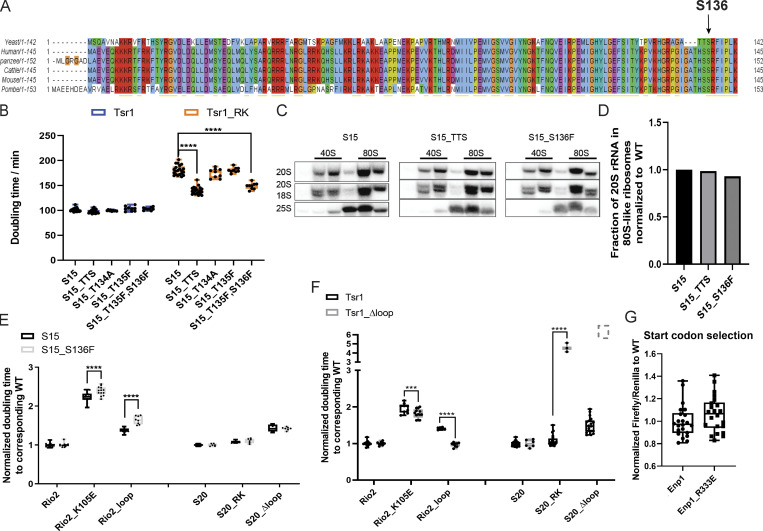
Conservation analysis of Rps15 (related to Fig. 7). (A) Rps15 sequences from the indicated organisms were aligned using the Clustal algorithm. The location of yeast S136 is indicated. (B) Doubling times of Gal:Tsr1,Gal:S15 cells supplemented with WT Tsr1 or Tsr1_RK and the indicated WT or CLL-associated Rps15 mutant plasmids. Significance was tested using a two-way ANOVA test. n ≥ 9; ****, P < 0.0001. (C) Northern blots of 10–50% sucrose gradients from Fap7-depleted cells supplied with WT S15 or S15_TTS or S15_S136F plasmids. (D) Normalized quantification of the data in C. Normalized doubling times of Gal:S15,Gal:Rio2 and Gal:S15,Gal:S20 cells supplied with S15 or S15_S136F and WT or mutant Rio2 plasmids and WT or mutant S20 plasmids. Significance was tested using a two-way ANOVA test. n ≥ 6, ****, P < 0.0001. (E and F) Normalized doubling times of Gal:Tsr1,Gal:Rio2 and Gal:Tsr1,Gal:S20 cells supplied with WT Tsr1 or Tsr1_Δloop and Rio2 plasmids (WT Rio2, Rio2_K105E, and Rio2_loop) or S20 plasmids(S20, S20_RK, S20_Δloop), respectively. Cells with Tsr1_Δloop and S20_Δloop do not grow and are therefore shown as a dotted box. Significance was tested using a two-way ANOVA test. n≥ 6 (except for Tsr1_ Δloop + S20_RK, n = 3); ***, P < 0.001; ****, P < 0.0001. (G) Fidelity of start-codon selection of Enp1_R333E relative to WT Enp1. Significance was tested using an unpaired t test. n = 22.
We then screened Rps15_TTS for rescue of the collection of mutants defective in the formation of 80S-like ribosomes. This screen reveals that Rps15_TTS rescues the defect from Tsr1_RK (Fig. S5 B). Further analysis demonstrates that the rescue of the Tsr1_RK growth defect arises from the Rps15_S136F mutation (Fig. S5 B), the residue most conserved between yeast and humans. This rescue is specific to the Tsr1_RK mutation; Rio2 mutants (Rio2_K105E, Rio2_loop) demonstrate small synthetic negative interactions with Rps15_S136F, while the Rps20 mutants are neutral (Fig. S5 E).
Relative to earlier pre40S intermediates, in 80S-like ribosomes, the C-terminal domain of Tsr1, which contains Tsr1_RK, is unlinked from the head and shifted toward the beak (Rai et al., 2020 Preprint). While this involves a rigid-body movement around the N-terminal extension of Tsr1 (Fig. S1 A; Rai et al., 2020 Preprint), we wondered if it was also accompanied by conformational changes in Tsr1 that could compensate for the lost binding interactions with Rps15, Ltv1, and Rio2. Because no such changes were visible in the parts of Tsr1 that are solved in the crystal and EM structures, we therefore focused on the parts of Tsr1 deleted from the protein in order to crystallize it successfully (McCaughan et al., 2016). As they prevent crystallization and are invisible in the EM structures, they appear to be mobile. The most notable such unresolved region is a large loop of Tsr1 between amino acids 410 and 476.
Deletion of this loop (Tsr1Δloop) has no effect on cell growth in a WT background (Fig. 7 B). In contrast, loop deletion in the context of the Tsr1_RK mutation rescues growth to near-WT levels (Fig. 7 B). As observed with the Rps15_S136F mutation, rescue is limited to Tsr1_RK and not observed with the Rio2 mutations, although Rio2_K105E and Rio2_loop are epistatic (Fig. S5 F). Rps20_RK and Rps20_Δloop are strongly synthetically sick (Fig. S5 F). Thus, other than the rescue of Tsr1_RK, these effects are opposite to the effects observed from Rps15_S136F.
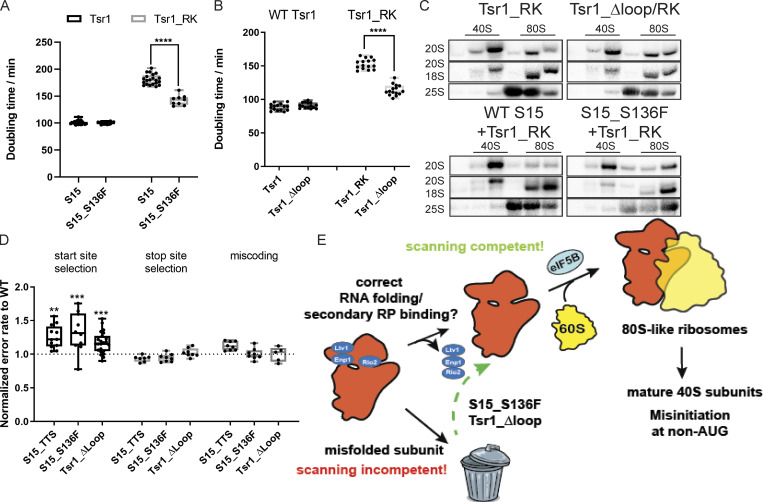
Bypass of quality control leads to defective start-codon recognition. (A) Doubling times of Gal:Tsr1,Gal:S15 cells supplemented with plasmids encoding WT Tsr1 or Tsr1_RK and WT S15 or the CLL-associated Rps15_S136F. The location of Rps15_S136F is highlighted in sphere in Fig. 5 A. Significance was tested using a one-way ANOVA test. n ≥ 9; ****, P < 0.0001. (B) Doubling times of Gal:Tsr1 cells supplemented with Tsr1 or Tsr1_ Δloop or Tsr1_RK or Tsr1_Δloop/RK plasmids. Significance was tested using an unpaired t test. n = 15; ****, P < 0.0001. (C) Northern blots of 10–50% sucrose gradients from Fap7 depleted cells expressing only the indicated Tsr1 and Rps15 mutants. Note that the blot for Tsr1_RK is the same as in Fig. 1 C. (D) Dual-luciferase translational fidelity assay of cells expressing S15_TTS, S15_S136F, or Tsr1_ Δloop. Significance was tested using an unpaired t test. n ≥ 5; **, P < 0.01; ***, P < 0.001. (E) Model for quality control of head assembly and scanning competence during formation of 80S-like ribosome assembly intermediates. Bypass of this mechanism leads to defective start-codon selection.
Together, these genetic interactions indicate that there are at least three distinct events that link Ltv1 phosphorylation, Rio2 phosphorylation, and Ltv1 release. One is blocked by the Rps20 mutants, one is blocked by Rio2 mutants, and one is blocked by Tsr1_RK. Previous data indicate that Rps20_Δloop blocks Rio2 phosphorylation, while K105E does not (Ferreira-Cerca et al., 2012; Mitterer et al., 2019), indicating that the Rps20 mutations affect communication from phosphor-Ltv1 to Rio2, while the Rio2 mutations affect communication from phosphorylation of Rio2 to release Ltv1 (Fig. 1 E). The observation that the communication between the same two proteins (Rio2 and Ltv1) requires different proteins in each case (Rps20 or Rio2) indicates that Rio2 phosphorylation is accompanied by a conformational change in Rio2, the pre40S or both.
Sucrose-gradient analysis demonstrates that both Rps15_S136F and the Tsr1_Δloop also rescue the block in the formation of 80S-like ribosomes observed in the Tsr1_RK mutation (Fig. 7 C). Thus, these mutations allow for bypass of the maturation defects that arise from the Tsr1_RK mutation, thereby rescuing growth.
h31 bypass mutants have defects in start-codon recognition
Having shown that Tsr1_Δloop and Rps15_S136F bypass the test for the integrity of h31 folding, we next tested if bypass of this quality control step affected the functionality of the 40S subunit during translation. To assess this, we used previously described dual-luciferase reporter plasmids that measure the frequency of mistakes in selection of start and stop codons and decoding. Expression of firefly luciferase from these plasmids relies on a mistranslation event (initiation at a UUG, stop codon readthrough, or misreading an arginine codon for histidine, respectively) and is normalized to expression of renilla luciferase to account for general translation defects, artifacts during extract preparation, etc. This analysis demonstrates that both Tsr1Δloop and Rps15_S136F have small but statistically significant defects in start-codon recognition, while the other steps in translation are not affected (Fig. 7 D). Of note, this analysis is done without perturbation of 40S maturation by the Tsr1_RK mutation. Similarly, Enp1_R333E (Fig. S5 G) and ΔLtv1 (Collins et al., 2018), which bypass the release of Enp1 and Ltv1, respectively, also display small defects in start-codon selection.
Thus, these mutations, which bypass the block in the formation of 80S-like ribosomes from mutants that perturb Ltv1 or Enp1 release, promoting the formation of 80S-like ribosomes and thereby rescuing growth defects, have deficiencies in the fidelity of start-codon selection. In contrast, A1193U, which impairs the formation of 80S-like ribosomes, displays more stringent AUG selection (Nemoto et al., 2010). Together, these data demonstrate that the formation of 80S-like ribosomes is a quality control step that probes proper assembly of the head to ensure that nascent subunits are competent for correct start-codon selection.
Discussion
Together, the two ribosomal subunits contain 4 rRNAs and 79 RPs. While some RPs play major structural roles and are required for subsequent assembly steps, others are small, located more peripherally, and incorporated late. Therefore, failure to assemble these proteins into ribosomes is not expected to globally disrupt assembly, and thus, ensuring their incorporation into every ribosome is not trivial. Nonetheless, deletion of even the smallest proteins produces growth or fidelity defects (Collins et al., 2018; Ferreira-Cerca et al., 2005; Pöll et al., 2009), indicating their importance for the functional integrity of the subunit.
To facilitate quality control of the assembly of nascent 40S subunits, cells use the translational machinery in a test drive, where the pre40S are joined by mature 60S subunits to form 80S-like ribosomes (Lebaron et al., 2012; Strunk et al., 2012). Within 80S-like ribosomes, the ATPase Fap7 tests the ability of the nascent small subunits to adopt the rotated state, a critical intermediate during translocation. Bypass of this step leads to defects in reading frame maintenance during translation (Ghalei et al., 2017). This work describes how the formation of 80S-like ribosomes tests a second function of the small subunit, the ability to properly identify a start codon, by ensuring the relevant conformations can be readily adopted.
80S-like ribosomes resemble the scanning-competent complex
Translation initiation requires the 40S subunit to bind an mRNA at the cap and then scan it to identify a start codon. This triggers a set of rearrangements, whereby the head closes around the mRNA•tRNA match, ultimately leading to the eIF5B-dependent formation of 80S complexes (Hussain et al., 2014; Llácer et al., 2015). A large body of biochemical data has demonstrated that mutations that affect start-codon fidelity disturb the equilibrium between the scanning competent and the closed conformation of the 40S subunit. Thus, the ability to form the scanning-competent complex is critical for correct start-codon selection, with mutations that destabilize this complex leading to misinitiation at non-AUG codons (Dong et al., 2017; Fekete et al., 2007; Hinnebusch, 2017; Martin-Marcos et al., 2017; Obayashi et al., 2017; Saini et al., 2010).
Comparison of the structure of nascent 40S subunits in 40S and 80S-like ribosomes with the subunits in scanning-competent and closed initiation complexes indicates that 80S-like ribosomes resemble scanning-competent complexes while the earlier pre40S subunits resemble the closed complex (Fig. 1 A). The data here demonstrate how this similarity is used to quality test the ability to correctly adopt this structure, ultimately ensuring the fidelity of start-codon selection (Fig. 7 E).
The formation of 80S-like ribosomes requires correct rRNA folding and assembly of RPs into the 40S subunit head
The data herein (Fig. 6 A) demonstrate that the formation of 80S-like ribosomes, whose structure resembles the scanning-competent complex, requires the step-wise phosphorylation and dissociation of Ltv1, Enp1, and Rio2. These steps are communicated to the next event through the 40S subunit head, which binds these AFs, in ways that requires and thus tests the correct folding of h31 and j34, two late-folding elements of the small ribosomal subunit. In addition, these events also require the correct binding and positioning of Rps3, Rps15, Rps20, and Rps29, universally conserved RPs in the head. Misfolding of the RNA, via the introduction of rRNA mutations, or deletion of the rRNA chaperone Ltv1, impairs the formation of 80S-like ribosomes. Thus, the formation of 80S-like ribosomes tests the correct formation of rRNA structure and RP incorporation across the subunit head, allowing progress only after each individual assembly step has been completed. In addition, our data also indicate that intermediates that cannot mature are instead degraded.
Bypass of individual test points leads to defective translation initiation
Our data demonstrate that the cancer-associated Rps15_S136F mutation, the Tsr1Δloop deletion, and the Enp1_R333E mutation each do not have any effects on growth or subunit joining by themselves but instead rescue the growth and subunit joining defects from the Tsr1_RK mutation or mutations that destabilize h34 (Rps20_DE, Rps30_EYER, and Rps3_KK). Furthermore, reporter assays for translational fidelity show that ribosomes from the Rps15_S136F, Tsr1Δloop, and Enp1_R333E mutants, or the Ltv1 deletion, have defects specifically in start-codon selection but do not affect general decoding or stop codon recognition. We thus suggest that the fidelity defects arise because these mutants, which bypass the checkpoints for correct head assembly, allow for the release of defective ribosomes into the translating pool, which would otherwise be degraded.
Taken together, these data demonstrate that formation of 80S-like ribosomes during 40S assembly is a quality control mechanism that tests the correct folding of head rRNA and its binding to RPs. Because the conformation of the head in 80S-like ribosomes resembles the structure in scanning-competent complexes, these assembly steps also test the ability to adopt this structure, thereby ensuring the scanning competence of the nascent 40S and its ability to discriminate against non-AUG start codons.
Does bypass of scanning competence checkpoint drive leukemia?
Approximately 20% of all cases of CLL display mutations in the C-terminal extension of Rps15 (Bretones et al., 2018; Ljungström et al., 2016). While it has been assumed that these mutations affect the function of Rps15 during translation, our results in yeast suggest that the cancer-promoting effects could also stem at least in part from the bypass of 40S quality control steps and thus be ultimately reflective of misassembled ribosomes. While we cannot rule out that the Rps15_S136F mutation has a direct effect on translation, our data show that in addition (or instead), it affects quality control during 40S maturation. The data furthermore indicate that the start-codon recognition defects from the Rps15_S136F mutation are a result of assembly defects, because their effects closely parallel the effects from the Tsr1Δloop mutation; neither of these affect growth or subunit joining in an otherwise WT background, both rescue the 40S assembly defects of the Tsr1_RK mutation, and both have genetic interactions with the Rio2 and Rps20 mutations. Moreover, both produce the same effects on start-codon selection. Because Tsr1 is not part of mature ribosomes, the effects from Tsr1Δloop must arise during assembly. Similarly, Enp1_R333E, which rescues the Enp1 release defects from a variety of mutants around j34, also demonstrates fidelity defects. Again, Enp1 is not present in mature ribosomes and must exert its defect during assembly.
How could the Rps15_S136F mutation promote cancer in human cells? Increased misinitiation at non-AUG codons is similarly observed with the L96P mutation in eIF1. Recent genome-wide profiling studies demonstrate that eIF1_L96P perturbs translation genome-wide, not just for the two yeast genes with a non-AUG start codon (Zhou et al., 2020). As a result, genes with a poor Kozak context are up-regulated. Furthermore, the utilization of upstream ORFs is increased, as these tend to have non-AUG codons or poor Kozak context. This affects translation of the downstream gene (often negatively). Thus, the data demonstrate a global reprogramming of translation in this mutant, and we presume that a similar reprogramming underlies the cancer-promoting effect from the Rps15_S136F mutation.
Nonetheless, cancer-associated mutations in eIF1A suppress misinitiation (Martin-Marcos et al., 2017). Similarly, the eIF5 antagonist 5MP drives colorectal cancer by suppressing non-AUG initiation, producing an oncogenic Myc isoform (Sato et al., 2019; Tang et al., 2017). Thus, misinitiation at non-AUG codons can also be tumor suppressive. These opposing effects can be explained by the presence of different transcripts (or transcript isoforms) in different cell types, and might underlie the observed cancer-tissue specificity for these mutations (CLL for Rps15 mutations, uveal melanoma for eIF1A mutations, and colorectal cancer for 5MP).
Our data also show that the formation of 80S-like ribosomes ensures the correct binding of Rps3, Rps15, Rps20, and Rps29. Furthermore, Ltv1-deficient yeast and human cells, which bypass the checkpoints for Ltv1 phosphorylation and release and are therefore not checked for correct binding of Rps3, Rps15, Rps20, and Rps29, have reduced levels of Rps10, Asc1, and possibly Rps29 in their ribosomes, likely due to mispositioning of Rps3 (Collins et al., 2018). Recent studies in cancer cells have demonstrated that these lose the stoichiometry of their RP composition (Ajore et al., 2017; Collins et al., 2018; Guimaraes and Zavolan, 2016; Kulkarni et al., 2017) and that this is associated with a poor outcome. Thus, the cancer-promoting effects from the bypass mutations may also arise, at least in part, from a loss in the stoichiometry of these RPs.
The small subunit head is prone to misfolding
During translation, the small ribosomal subunit carries out three important functions: (1) during initiation it, identifies the correct start codon, thereby setting the reading frame while also directing the synthesis of the correct amount of protein from each mRNA; and (2) during elongation, it inspects the correct match between mRNA and tRNA while also (3) ensuring reading frame maintenance during translocation. Each of these functions is associated with conformational changes in the subunit, which are modulated by the binding of its ligands: translation factors and tRNAs (Munro et al., 2009). Thus, the small subunit rRNA sequence encodes not just one but multiple structures, whose equilibrium must be carefully balanced in order to ensure the fidelity of each of these processes. The most mobile portion of the subunit is the head structure, which can adopt multiple orientations in relationship to the body, the platform, and the large subunit.
Perhaps not coincidentally, the head structure is also the slowest to fold and assemble, likely because it is prone to misfolding (Adilakshmi et al., 2008; Duss et al., 2019). Intriguingly, the small subunit head has also seen the most dramatic changes during evolution, with parts of the beak rRNA replaced by RPs in the transition from bacteria to eukaryotes. Even more dramatically, in mitochondria the rRNA is substantially truncated and mostly replaced by RPs. Presumably, these adaptations facilitate correct folding and prevent misfolding of the structure. Despite these adaptations to the head, a common core remains, consisting of the universally conserved proteins uS7 (Rps5, missing in trypanosomal mitochondrial small subunit [mito-SSU]), uS9 (Rps16), and uS13 (Rps18, missing in trypanosomal mito-SSU), which bind cotranscriptionally, as well as uS3 (Rps3), uS19 (Rps15), uS10 (Rps20), and uS14 (Rps29), which bind in a second wave. Thus, the middle-binding Rps3, Rps15, Rps20, and Rps29 are conserved in all forms of life, demonstrating their importance for small subunit structure and/or function. This work describes a quality control mechanism evolved in eukaryotes to ensure their proper incorporation into a complex structure prone to misfolding. Whether analogous mechanisms exist to ensure their proper incorporation into bacterial or mitochondrial ribosomes remains unknown, but the bacterial AF RimM appears to have taken over at least some functions of Ltv1 (Collins et al., 2018). Similarly, a cluster of nine mito-AFs is located near uS3 in the trypanosomal assembly intermediate (Saurer et al., 2019). It is conceivable that these carry out some of the roles of Enp1 and Ltv1 described here.
Materials and methods
Plasmids and yeast strains
Yeast strains (Table S1) were generated by PCR-mediated homologous recombination and confirmed by PCR, serial dilution, and Western blotting if antibodies were available. Plasmids (Table S2) were constructed using standard cloning techniques and confirmed via sequencing.
Growth curve measurements
Cells were grown in YPD or glucose minimal media (if supplemented plasmid is not essential) overnight and then diluted into fresh YPD for 3–6 h before inoculating into 96-well plates at a starting OD600 between 0.04 and 0.1. The Synergy.2 plate reader (BioTek) was used to record OD600 for 48 h while shaking plate at 30°C unless otherwise specified. rRNA mutants were grown in minimal media at 30°C overnight and diluted into fresh medium for 3 h before shifting to 37°C for 3 h. Cells were then inoculated into YPD and the growth curve measured at 37°C. Doubling times were calculated using data points within the mid-log phase. Data were averaged from at least six biological replicates of three different colonies and two independent measurements.
In vivo subunit joining assay
Cells were grown to mid-log phase in galactose before inoculating into YPD for at least 16 h to deplete Fap7. Formation of 80S-like ribosomes was assayed by sucrose-gradient fractionation and Northern blotting as previously described (Ghalei et al., 2015; Strunk et al., 2012), and the fraction of 20S rRNA in 80S-like ribosomes was quantified. In brief, 5,000 OD of clarified lysate was loaded onto a 10–50% sucrose gradient, spun for 2 h at 40,000 rpm in an SW41Ti rotor, and fractionated into fractions of ∼700 µl. For Northern blotting, 200 µl of each fraction were phenol chloroform extracted. Dissociation of AFs was assayed by Western blotting, and the fraction of each AF in pre40S ribosomes was quantified. Cellular fractionation for ΔLtv1 cells was performed exactly as previously described (Ghalei et al., 2015). The effects of rRNA mutants were tested in Gal::Fap7 cells using the unique 18S tag sequence (Table S3) on the plasmid-derived RNA (Jeeninga et al., 1997). At least two biological replicates were assayed for each mutant (Table S4).
Dual-luciferase assay
Dual-luciferase assays were performed using the Dual-Luciferase Reporter Assay (Promega) as previously described (Ghalei et al., 2017). Specifically, 2 ml cells at mid-log phase was pelleted, washed, and flash frozen. Cells were resuspended in 1 ml passive lysis buffer and incubated at room temperature for 5 min. 10 µl lysate and 20 µl Luciferase Assay Reagent II were added to each well in black Fluotrac600 96W Microplates (Greiner Bio-One) to assay firefly Luciferase activity, followed by addition of 20 µl Stop&Glo Reagent to measure Renilla luciferase in a Centro LB 960 Microplate Luminometer. For each sample, Firefly activity was first normalized to Renilla activity before normalizing the firefly/Renilla ratio for each mutant to that for WT. Data were derived from at least eight biological replicates, with three technical replicates each.
Northern blotting
Northern blots were performed on total RNA from NOY 504 strains supplied with plasmids encoding WT 18S or RNA mutants. Cells were grown overnight in minimal glucose media at 30°C, diluted, and regrown to mid-log phase before inoculating to YPD. The cells were then grown at 37°C for at least 16 h to deplete genomically encoded rRNA. 18S-TAG was probed to make sure the RNA encoding plasmid persists in cells. 20S rRNA were normalized to 18S and 25S for comparison.
Quantification and statistical analysis
Western blots were imaged using the ChemiDoc MP Imaging System from Bio-Rad after applying luminescence substrates. Northern blots were visualized by Typhoon FLA 7000 (GE Healthcare). Image Lab (Bio-Rad) and Quantity One (Bio-Rad) were used to analyze Western blots and Northern blots, respectively.
Standard statistical tests were used as indicated in the respective figure legends and performed using Prism version 8.3 (GraphPad Software).
Online supplemental material
Fig. S1 is related to Fig. 1 and shows mutant doubling times, sample gradients, and the location of Northern probes. Fig. S2 is related to Fig. 4 and shows doubling times for S3 and S3_KK, S20, and S20_DE in the WT Ltv1 and Ltv1_S/D backgrounds, and a characterization of the Enp1_R333E mutant. Fig. S3 is related to Fig. 5 and shows a characterization of the Rio2_RQQR mutant. Fig. S4 is related to Fig. 6 and shows nuclear fractionation as well as the location and characterization of j34 rRNA mutants. Fig. S5 is related to Fig. 7 and shows the location and analysis of the Rps15_S136F mutant. Table S1 lists the yeast strains used in this study. Table S2 lists the plasmids used in this study. Table S3 lists the oligos used in this study. Table S4 lists the yeast strains and plasmids used in each figure.
Acknowledgments
The dual-luciferase plasmids were kindly provided by J. Lorsch (National Institutes of Health, Bethesda, MD; start site recognition) and D. Bedwell (University of Alabama at Birmingham, Birmingham, AL; stop codon readthrough and miscoding). We thank E. Horgen and N. Matese for help with generating plasmids and members of the Karbstein laboratory for discussion and comments on the manuscript.
This work was supported by National Institutes of Health grants R01-GM086451 and R01-GM117093 and Howard Hughes Medical Institute faculty scholar grant 55108536 (to K. Karbstein).
The authors declare no competing financial interests.
Author contributions: H. Huang, H. Ghalei, and K. Karbstein designed and performed the experiments. H. Huang and K. Karbstein wrote the paper.