
Tang et al. characterize a novel proteolysis and proteasome-dependent pathway involved in degradation of unfolded antibody heavy chain (HC). Unfolded HC is ubiquitinated by N-recognin E3 ligases UBR4 and UBR5, then extracted and degraded by the proteasome. The ER luminal protease PDIA3 cleaves ubiquitinated HC to accelerate HC dislocation.
Accumulation of unfolded antibody chains in the ER triggers ER stress that may lead to reduced productivity in therapeutic antibody manufacturing processes. We identified UBR4 and UBR5 as ubiquitin E3 ligases involved in HC ER-associated degradation. Knockdown of UBR4 and UBR5 resulted in intracellular accumulation, enhanced secretion, and reduced ubiquitination of HC. In concert with these E3 ligases, PDIA3 was shown to cleave ubiquitinated HC molecules to accelerate HC dislocation. Interestingly, UBR5, and to a lesser degree UBR4, were down-regulated as cellular demand for antibody expression increased in CHO cells during the production phase, or in plasma B cells. Reducing UBR4/UBR5 expression before the production phase increased antibody productivity in CHO cells, possibly by redirecting antibody molecules from degradation to secretion. Altogether we have characterized a novel proteolysis/proteasome-dependent pathway involved in degradation of unfolded antibody HC. Proteins characterized in this pathway may be novel targets for CHO cell engineering.
Therapeutic mAbs are used to treat a wide range of human diseases (Adams and Weiner, 2005). While eukaryotic expression systems are optimal for expression of mAbs, folding and assembly of newly synthesized antibody chains in the ER can be rate and yield limiting (Khan and Schröder, 2008; Nishimiya, 2014). When antibody synthesis rate exceeds ER capacity, the unfolded protein response (UPR) pathway is activated to halt protein translation, increase unfolded protein degradation, and improve protein folding by elevating expression of protein chaperones (Cenci and Sitia, 2007; van Anken et al., 2003). While correctly folded proteins proceed through the secretory pathway and are secreted outside of the cell, unfolded or misfolded proteins are directed toward ER-associated degradation (ERAD). ERAD involves substrate recognition, dislocation across the lipid bilayer to the cytosol, ubiquitination, and proteasomal degradation (Ruggiano et al., 2014). In the cytosol, ER-associated ubiquitin E3 ligases interact with, either directly or with the aid of luminal adaptors, and ubiquitinate ERAD substrates. Extraction of luminal domains of proteins targeted for degradation requires a protein-conducting channel or “dislocon,” the identity of which still remains controversial (Bagola et al., 2011; Ruggiano et al., 2014; Stein et al., 2014). Driving force and directionality of this process are thought to be mediated either by p97 (an AAA+ ATPase; Oberdorf et al., 2006; Ye et al., 2004) or the six proteasomal AAA ATPases at the ring base of the 19S regulatory particle (Bar-Nun and Glickman, 2012; Lee et al., 2004; Mayer et al., 1998; Oberdorf et al., 2006).
While most misfolded ER proteins are degraded by ERAD, a subset of misfolded secretory proteins are degraded through proteasome-independent mechanisms such as autophagy (Kamimoto et al., 2006; Perlmutter, 2006). Proteases also operate in a parallel manner to alleviate ER stress (Schmitz and Herzog, 2004). ER serine and/or cysteine proteases have been implicated in proteasome-independent ER protein degradation. For example, a protein known as ER-60 as well as protein disulfide isomerase (PDI) PDIA3 or ERp57, which is a calnexin-associated protein, have been suggested to be involved in degradation of misfolded human lysozyme mutant and hepatic apolipoprotein B100 (Otsu et al., 1995; Rutledge et al., 2013). Other proteases, including signal peptide peptidase (SPP) and rhomboid family protein RHBDL4, have been found to play roles in dislocation and degradation of transmembrane proteins in the ER (Boname et al., 2014; Chen et al., 2014; Fleig et al., 2012; Loureiro et al., 2006). Besides a few model substrates such as the major histocompatibility class molecules or Ig light chain (LC) molecules, the degradation of many secreted or membrane proteins, including IgG heavy chain (HC), is less characterized. It has been shown that SEL1L (an ER adaptor protein for the ERAD ubiquitin ligase Hrd1) and Hrd1 are involved in the degradation of μ HC (IgM HC; Cattaneo et al., 2008), and a truncated version of γ HC (IgG HC) that only contains VH and CH1 domains was ubiquitinated and degraded in the cell (Shimizu et al., 2010). In this study, we determined that two E3 ligases, UBR4 and UBR5, are involved in IgG HC ubiquitination and its subsequent proteasome-mediated degradation. Additionally, we have shown that the protease PDIA3/ER-60 cleaves unfolded antibody HC molecules, accelerating the dislocation of the ubiquitinated N-terminal domain of HC for degradation and making the remaining C-terminal domain available for another round of ubiquitination by UBR4/UBR5. Proteins involved in this degradation pathway may be potential targets for cell line engineering purposes.
To determine the mechanism of IgG HC degradation in CHO cells, we generated cell lines that stably expressed IgG HC fused with a C-terminal Flag tag, with or without the coexpression of LC fused with a C-terminal HA tag (Fig. S1 A). The proper folding of HC requires LC assistance (Feige et al., 2009), without which HC is expected to remain unfolded and be degraded. The expression of the HC molecule alone was unstable in our studies and was silenced within 3 mo, either due to transcriptional silencing (mAb2 HC, clone 1) or loss of HC gene copies (mAb2 HC, clone 2; Fig. S1, D–F; Kim et al., 2011). To determine whether the intracellular unfolded HC is mainly degraded by the ERAD pathway or autophagy, we tested the effects of proteasomal or lysosomal inhibitors in HC degradation. Treatment with the proteasomal inhibitor MG132 but not lysosomal inhibitors, such as choloroquine, NH4Cl, and BaflomycinA1, blocked HC degradation, indicating that HC is mainly subjected to proteasome-dependent ERAD, irrespective of its glycosylation state (Fig. 1 A). It has been suggested that retrotranslocation of some proteins including the unassembled IgG LC is tightly coupled to proteasome activity (Chillarón and Haas, 2000). To determine mode of HC degradation, we performed subcellular fractionation to separate the intracellular membranes from the cytosolic fraction in the presence or absence of MG132. We observed that HC and ubiquitinated HC were mainly detected in the membrane fractions (Fig. 1, B and C), suggesting that HC translocation and proteasomal degradation are also coupled.

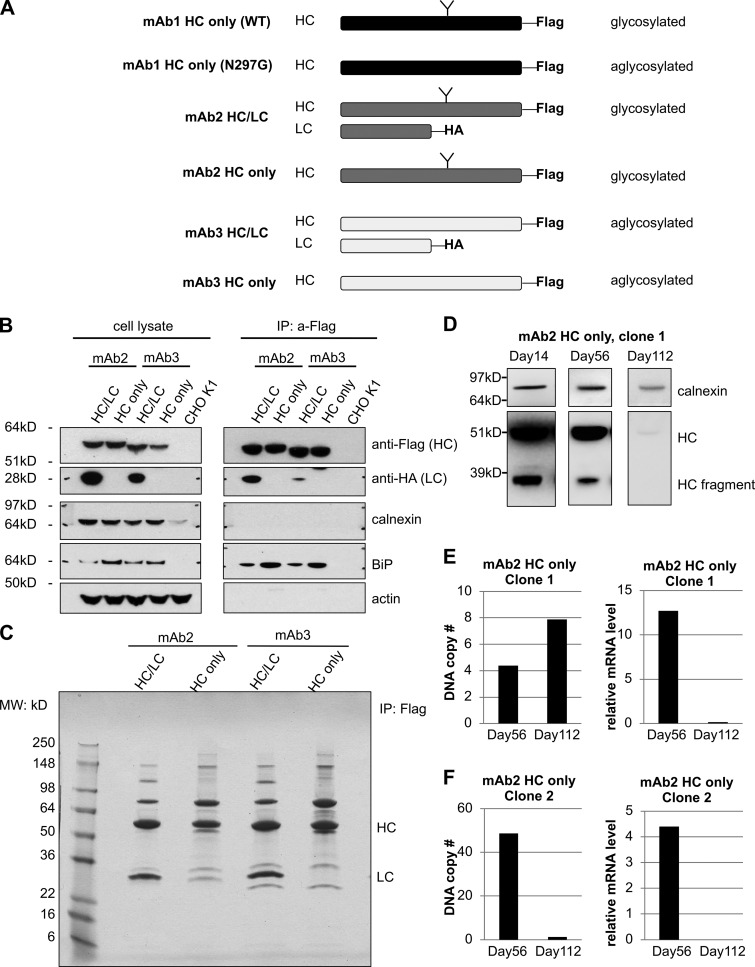
Identifying proteins that coimmunoprecipitate with the antibody HC. (A) Outline of Flag-tagged HC and HA-tagged LC constructs and the respective cell lines that were generated expressing different antibodies. (B) The levels of indicated proteins were analyzed in cell lysate (left) and HC immunoprecipitated samples (right) by Western blotting. (C) Antibody HC associating proteins from indicated cell lines were analyzed after immunoprecipitation by SDS-PAGE and Coomassie blue staining and used for mass spectrometry. (D) Levels of full-length HC and HC fragment in the whole-cell lysates of HC-expressing cells were analyzed by Western blotting after culturing them for the indicated durations. Calnexin was used as a loading control. (E and F) DNA copy number and relative mRNA levels of HC in two different clones at days 56 and 112. MW, molecular weight.
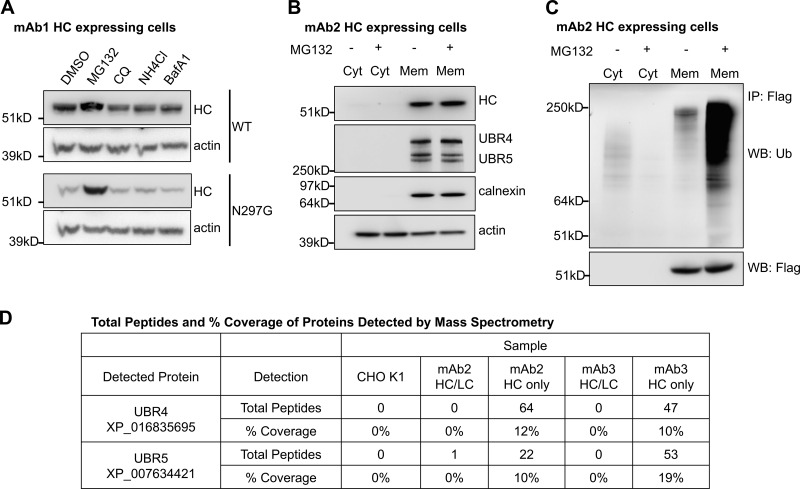
UBR4 and UBR5 localize to the ER membrane and associate with antibody HC. (A) Cells expressing WT or an aglycosylated mutant (N297G) version of antibody HC were treated with the indicated drugs for 20 h. The cells were then lysed, and the intracellular HC levels were analyzed. MG132 at 20 µM, CQ (chloroquine) at 50 µM, NH4Cl at 10 mM, and BafA1 (BafilomycinA1) at 1 µM. (B) Cells expressing antibody HC were treated with or without MG132 and subjected to subcellular fractionation. Subcellular localization of indicated proteins were analyzed by Western blotting. Cyt: cytosol, Mem: membrane fraction. (C) Antibody HC was immunoprecipitated from the cytosol and solubilized membrane fractions from 1B, and analyzed for ubiquitination via Western blotting. WB: antigen probed in Western blotting. IP: antigen used for immunoprecipitation. (D) Proteins that associated with antibody HC were coimmunoprecipitated, trypsinized, and analyzed by mass spectrometry. The table shows the number of total peptides and percentage of sequence coverage of the indicated proteins that were detected in each sample. Total peptides: total number of peptides generated from the indicated protein in each sample, which suggests the relative amount of the indicated protein. % Coverage: the percentage of the sequence of the indicated protein that was covered by identified peptides, where higher percentages suggest increased confidence of detection. HC only: cells that express only HC. HC/LC: cells that express both HC and LC.
To identify proteins that are involved in IgG ERAD, we performed a coimmunoprecipitation assay followed by mass spectrometry using cell lines that express HC alone or together with LC (Fig. S1, B and C; and Fig. 1 D). When expressed without LC, HC molecules are expected to be recruit to ERAD machinery more robustly. Two ubiquitin E3 ligases, UBR4 and UBR5, were identified by mass spectrometry to be coimmunoprecipitated with HC (Fig. 1 D). By mass spectrometry, it was harder to detect the association of UBR4 and UBR5 with IgG HC when coexpressed with LC as opposed to expression by itself. Nevertheless, this association was also confirmed by Western blotting for cells expressing complete IgG (Fig. S3 A). UBR4 and UBR5 belong to the mammalian ubiquitin protein ligase E3 component N-recognin (UBR) family (Tasaki et al., 2005), which recognize substrates for degradation in the N-degron pathway. The N-degron pathway relates the stability of a protein to the N-terminal residue of that protein (Dougan et al., 2012). In this pathway, destabilizing N-terminal residues of short-lived proteins act as degradation determinants (N-degrons). Substrates carrying N-degrons are recognized and ubiquitinated by UBR family proteins and degraded by proteasomes. Although UBR family proteins are mainly cytosolic E3 ligases, in yeast Ubr1 seems to be involved in the ERAD of two transmembrane ER client proteins (Stolz et al., 2013). Subcellular localization analysis confirmed that in IgG HC–expressing CHO cells, a subpopulation of UBR4 and UBR5 was localized on ER membranes (Fig. 1 B and Fig. S2), suggesting that they could be involved in the ERAD pathway.
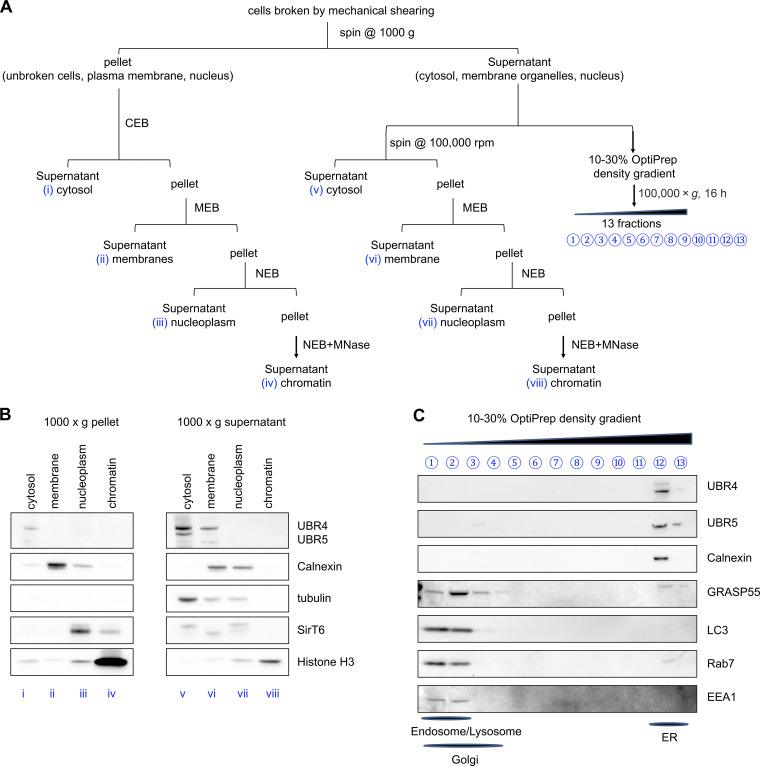
Subcellular localization of UBR4 and UBR5. (A) Schematic overview of the subcellular fractionation procedure. IgG-expressing CHO cells were homogenized by mechanical shear and centrifuged at 1,000 g for 10 min to pellet the intact cells, nuclei and plasma membranes (i–iv). The supernatant that contains cytosol and membrane organelles was then either subjected directly to ultracentrifugation at 100,000 g to separate cytosol (v) and pellet the organelles (vi–viii), or loaded onto an OptiPrep gradient for density separation (C). Both the pellet generated from 1,000 g (i–iv) and the pellet generated from 100,000 g (vi–viii) centrifugations were further fractionated by solubilizing proteins using Thermo Fisher Scientific Subcellular Protein Fractionation Kit for Cultured Cells (B). MEB, membrane extraction buffer; NEB, nuclear extraction buffer; MNase, micrococcal nuclease. (B) Western blot of indicated proteins from fractions i–viii. (C) Western blot of indicated proteins from OptiPrep gradient fractions 1–13. Established markers for cellular compartments used include ER marker calnexin, cytosol marker α-tubulin, nucleoplasm marker SirT6, chromatin marker histone H3, Golgi apparatus marker GRASP55, lysosome marker LC3, late endosome marker Rab7, and early endosome marker EEA1.
RNAi-mediated depletion of UBR4, UBR5, or both in HC-expressing cells increased intracellular levels of antibody HC (Fig. 2, A and B). Analysis of antibody HC levels in the intracellular membrane fraction revealed the presence of a 35-kD HC fragment, detected by both anti-Flag and anti-human IgG antibodies, and further experiments confirmed that HC molecules were cleaved in the ER and not after lysis (Fig. S3 B). These HC fragments were sometimes more readily detectible relative to the full-length HC in the membrane fraction when UBR4, UBR5, or both are depleted (Fig. 2 C). These results (Fig. 2, A–C) suggested that both UBR4 and UBR5, acting in a complex or sequentially, were involved in HC degradation. Since both UBR4 and UBR5 were found to be associated with antibody HC, we decided to knock down both of these proteins throughout our studies. To rule out clone-specific behaviors, UBR4 and UBR5 were simultaneously knocked down in eight different clones expressing four different HC molecules, including both glycosylated and aglycosylated versions (Fig. S4, A and B). In all cases, UBR4 and UBR5 double depletion caused intracellular accumulation of HC fragment. Additionally, depletion of both UBR4 and UBR5 in four different clones expressing two different full antibody molecules (both antibody HC and LC) resulted in accumulation of HC fragment inside the cell and increased the secretion of a high molecular weight species (HMWS) of HC outside of the cell (Fig. S4, C and D).
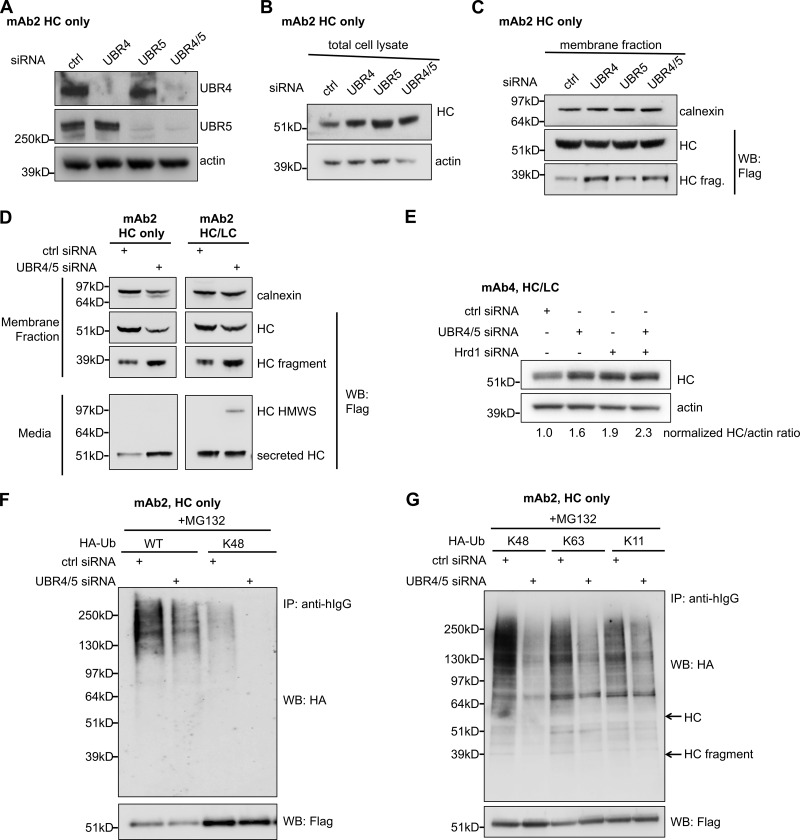
UBR4 and UBR5 ubiquitinate antibody HC and target it for degradation. (A) Knockdown of UBR4 and UBR5 by siRNA transfection. UBR4/5: UBR4 and UBR5. (B) Intracellular levels of HC in HC-expressing cells were determined after cells were treated with the indicated UBR siRNA oligos for 4 d. (C) Levels of full-length HC and HC fragment in the membrane fraction of HC-expressing cells treated with the indicated UBR siRNA oligos. (D) Pools of mAb2 HC/LC- or HC only–expressing cells were treated with control siRNA or both UBR4 and UBR5 specific siRNA oligos. Levels of full-length HC and HC fragment in both the intracellular membrane fraction and in the media (harvested cell culture fluid or HCCF) were analyzed by Western blotting. Calnexin (an ER protein) was used as loading control of intracellular membrane fractions. (E) Cells expressing mAb4 HC/LC were transfected with indicated siRNA(s) and subjected to Western blot analysis for HC and actin. Relative HC/actin band intensity ratios have been quantified for each sample and normalized to the control. (F) Cells that stably express Flag-tagged mAb2 HC and HA-tagged WT or K48-specific mutant ubiquitin were transfected with UBR4/5 or control siRNA oligos followed by MG132 treatment. HC molecules in these cells were then immunoprecipitated. The levels of HA-Ub modifications were determined by Western blotting. (G) Cells that stably express Flag-tagged mAb1 HC and HA-tagged K48, K63, or K11-specific ubiquitin mutants were treated as in F. HC molecules were immunoprecipitated, and HA-Ub modifications were determined by Western blotting. K48, K63, or K11-specific mutants of ubiquitin have all lysine residues mutated to alanine except for the indicated lysine residue. hIgG: human IgG. Arrows indicate the expected migration positions of HC and HC fragment bands on SDS-PAGE.
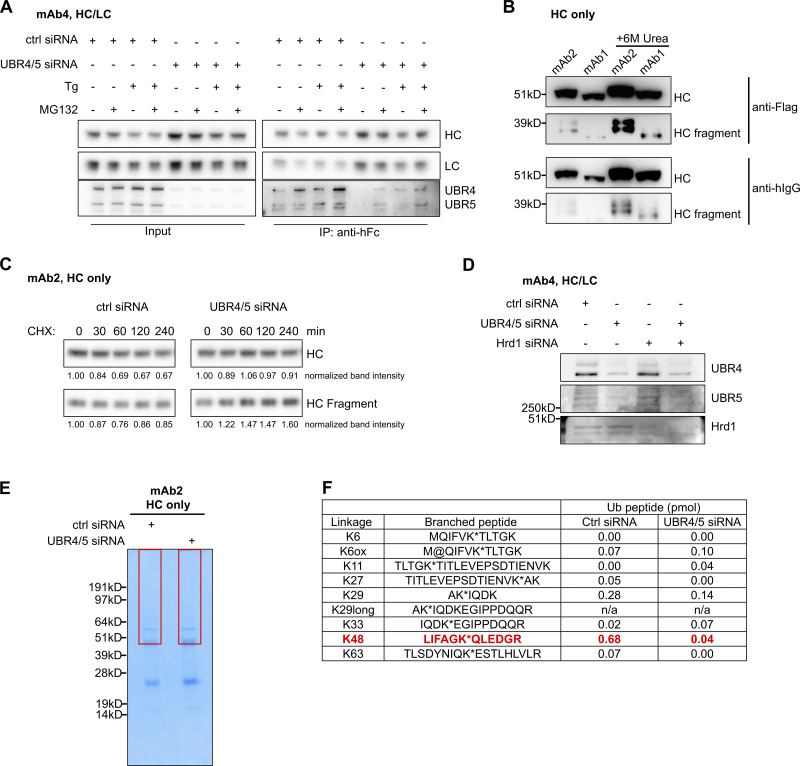
HC cleavage and ubiquitination in the cell. (A) Cells expressing mAb5 were transfected with either control or UBR4/UBR5 siRNA. In control siRNA–treated cells, UBR4 and UBR5 associated with IgG HC when HC was immunoprecipitated by anti-human Fc resins. This association was stronger when cells were treated with thapsigargin (Tg) and/or MG132 to induce ER stress. (B) Cells expressing mAb1 or mAb2 HC were lysed in the absence or presence of 6 M urea. HC and HC fragments were detected by Western blotting, using either anti-Flag or anti-human IgG antibodies. hIgG: human IgG. (C) Cells expressing mAb2 HC were transfected with control siRNA or UBR4 and UBR5 siRNA combination for 96 h. These cells were then treated with CHX to block protein synthesis. The intracellular levels of HC in the treated cells were probed by anti-human Fc antibodies. The levels of HC or HC fragment in each sample were quantified and normalized to the zero time point. Note that in control siRNA–treated cells, the level of intracellular HC reduced over time, while in UBR4/5– depleted cells, HC level was relatively stable. (D) Cells expressing mAb4 HC/LC were transfected with indicated siRNA(s) and subjected to Western blot analysis for indicated proteins to evaluate knockdown efficiencies. (E) Cells expressing mAb2 HC were transfected with control or UBR4/5 siRNAs. HC was immunoprecipitated from an equal amount of cell lysates, then eluted from anti-human Fc agarose by 6 M urea. Protein bands were separated by SDS-PAGE and visualized using Simply Blue staining. Regions above the 55-kD HC band (area in the boxes) to the top of the gel till the sample input well were excised and analyzed by mass spectrometry. (F) Amount of branched ubiquitin peptides determined in each sample of E. *, -GG linked; ox or @, oxidized methionine.
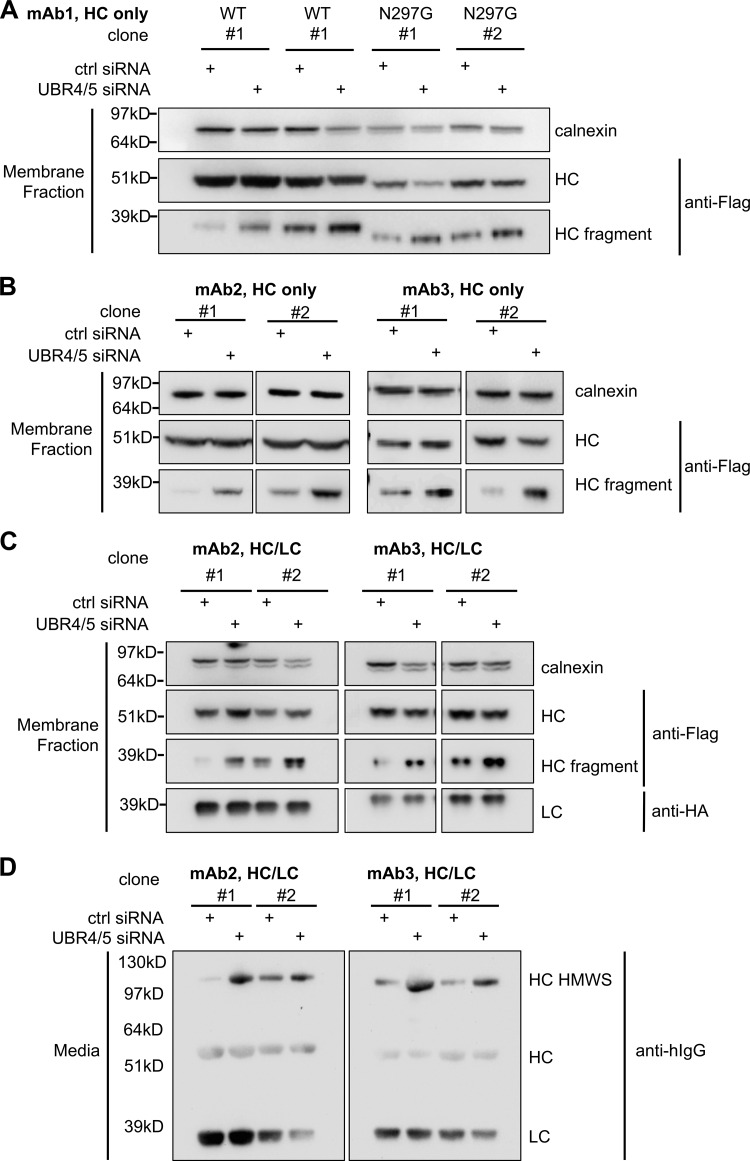
UBR4 and UBR5 are involved in HC degradation in both HC only– and full antibody–expressing clones. (A and B) Cells expressing indicated mAb HC were transfected with control siRNA or UBR4/5 siRNA. Levels of full-length HC and HC fragment in the membrane fractions of these cells were analyzed by Western blotting. (C and D) Cells expressing indicated mAb HC and LC were transfected with control or UBR4/5 siRNA. Levels of antibody HC and LC in the membrane fractions (C) and in the media (D) were analyzed by Western blotting.
To exclude clone variations, pools of cells expressing full IgG or the HC alone (mAb2) were generated (Fig. 2 D), and depletion of both UBR4 and UBR5 in these pools resulted in accumulation of HC fragment in the intracellular membrane fraction (Fig. 2 D, top panels). Additionally, secretion of intact HC or HMWS of HC was increased for HC only–expressing or full IgG–expressing cells, respectively (Fig. 2 D, bottom panels). These results indicate that even with LC coexpression, a subset of HC molecules remains unfolded and is eventually degraded (Fig. 2 D). Furthermore, in the pool of cells expressing HC alone, UBR4 and UBR5 depletion stabilized HC in the cycloheximide (CHX) chase assay, when total protein synthesis was blocked by CHX treatment (Fig. S3 C), further confirming that UBR4 and UBR5 are involved in HC degradation.
To determine whether other ERAD E3 ligases are involved in HC degradation, we depleted Hrd1 or gp78 by siRNA transfection in IgG-expressing CHO cells. Although gp78 depletion did not affect HC level (data not shown), Hrd1 depletion increased intracellular HC levels, similar to UBR4 and UBR5 depletion. Depletion of Hrd1 in addition to UBR4/5 depletion further increased intracellular HC levels (Fig. 2 E and Fig. S3 D), indicating that multiple E3 ligases are involved in HC degradation, and they may all ubiquitinate HC during this process.
To test whether UBR4 and UBR5 ubiquitinate IgG HC, we generated cell lines that stably expressed Flag-tagged IgG HC together with HA-tagged WT or lysine-specific ubiquitin (Ub) constructs (Misaghi et al., 2009). In the K48-specific ubiquitin mutant, all lysine residues have been mutated to alanine except for K48 to allow formation of K48-specific poly-ubiquitin chains only. Similarly, K63- and K11-specific ubiquitin mutants can only form K63- and K11-specific poly-ubiquitin chains, respectively. These cell lines were treated with control or UBR4 and UBR5 siRNA, and intracellular HC was immunoprecipitated after 20 h of MG132 treatment, using anti-Flag agarose beads. The ubiquitination level of HC was then analyzed by blotting for HA-Ub. Depletion of UBR4 and UBR5 reduced total HC ubiquitination, particularly K48 specific ubiquitination (Fig. 2 F), suggesting that UBR4 and UBR5 are required for HC ubiquitination. The more drastic reduction in the levels of K48 relative to other lysine-specific ubiquitinations (Fig. 2 G) suggests that UBR4 and UBR5 mainly catalyze K48 ubiquitination of HC molecules; however, this does not exclude possible involvement of other E3 ligases that might facilitate formation of mixed ubiquitin branches on IgG HC. Using mass spectrometry, we were able to determine branched -GG signature peptides that indicated the linkage of poly-Ub chains through different lysine residues (Phu et al., 2011; Fig. S3, E and F). The result showed that K48 linkage is the major linkage of HC ubiquitination, and depletion of UBR4 and UBR5 mainly reduced K48-linked chains.
Accumulation of the intracellular HC fragment in the membrane fraction, upon UBR4 and UBR5 depletion (Fig. 2, C and D; and Fig. S4), prompted us to investigate the role of IgG HC cleavage in its degradation. The cleaved HC fragment represented the C-terminal portion of this molecule, as it could be recognized by anti-flag antibody (Fig. S3 B). N-terminal sequencing determined that the cleavage occurred between the asparagine 211 and threonine 212 residues (Fig. 3 A) and identically so as evaluated for three different IgG1 molecules. Based on molecular modeling, the site of HC cleavage was mapped to an exposed loop on the surface of the HC molecule (Fig. 3 B). RNAi screening of seven well-established proteases in the secretory pathway revealed that only PDIA3 depletion prevented IgG HC cleavage (Fig. 3 C). Additionally, reanalysis of the mass spectrometry data of the co-immunoprecipitation experiment (Fig. S1 C) showed that PDIA3 might interact with assembled antibody and HC molecules, indicative of their potential association in the ER (Fig. 3 D).
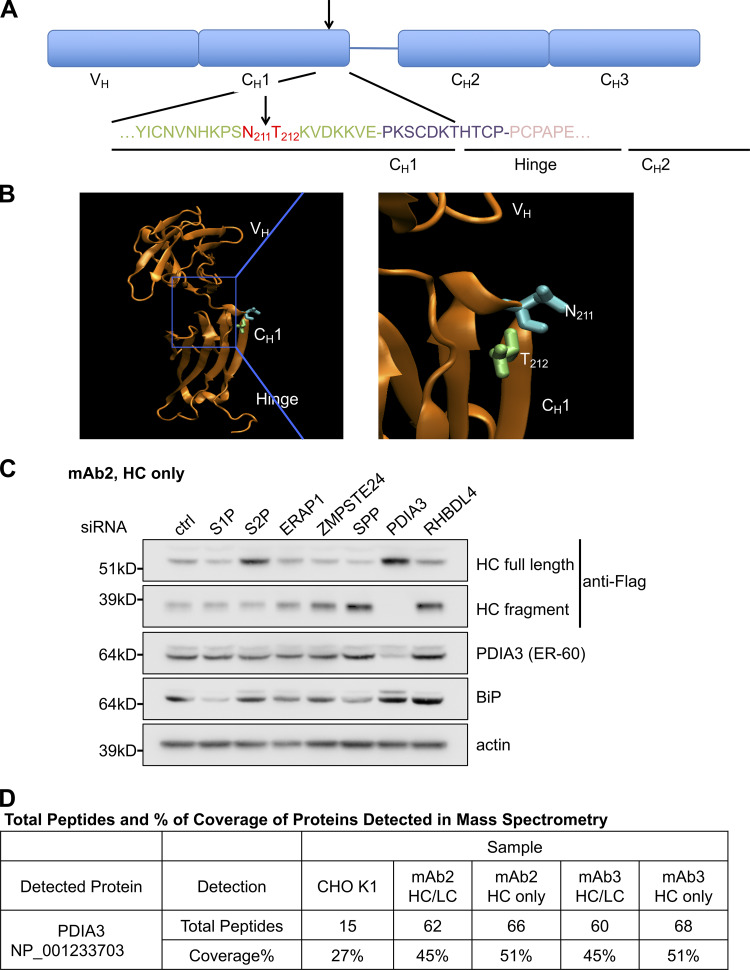
PDIA3 is involved in antibody HC cleavage. (A) The cleavage site of antibody HC was determined by N-terminal sequencing of the C-terminal HC fragment. The cleavage site for all the analyzed HC molecules was determined to be between asparagine 211 and threonine 212. Arrow indicates the cleavage site. (B) The cleavage site of antibody HC is in an exposed helix as shown in the ribbon diagram of the protein structure. (C) Cells expressing mAb1 HC were transfected with siRNA oligos specific to the indicated genes. Levels of full-length HC, HC fragment, and other indicated proteins were analyzed by Western blotting. (D) PDIA3 was identified as one of the proteins that associated with antibody HC or fully assembled antibody (HC/LC) by mass spectrometry as in Fig. 1 A.
PDIA3, also known as ER-60, has both protein disulfide isomerase and cysteine protease activities and is known to be involved in hZLM and apoB cleavage and degradation (Otsu et al., 1995; Rutledge et al., 2013). PDIA3 has two CGHC motifs that are the active sites of both its cysteine protease and isomerase activities. Replacing the second cysteine in both CGHC motifs to alanine (C60A/C409A, CGHA mutant) abolishes the protease activity and reduces the isomerase activity of PDIA3, while mutating the first cysteine in both CGHC motifs to alanine (C57A/C406A, AGHC mutant) only eliminates the isomerase activity (Kikuchi et al., 2002; Rutledge et al., 2013; Urade et al., 1997). To validate that the cysteine protease and not the isomerase activity of the PDIA3 is required for HC cleavage, we exogenously expressed WT or indicated mutants of PDIA3 in the WT-background IgG-expressing cells. The expression of exogenous PDIA3 was confirmed by knocking down the endogenous protein by siRNA in Fig. S5 A. As shown in Fig. 4 A, expression of the CGHA but not the AGHC mutant inhibited HC cleavage, suggesting that only cysteine protease activity was required for PDIA3 to cleave IgG HC. Furthermore, HC cleavage was inhibited by a cysteine protease inhibitor (p-hydroxymercuribenzoic acid sodium salt [pHMB]) but not by a proteasomal or lysosomal protease inhibitor (ALLN; Fig. 4 B; Qiu et al., 2004; Rutledge et al., 2013). In a CHX chase experiment, when the cells were treated with CHX to block protein synthesis, we were able to track the cleavage of HC and the degradation of HC and HC fragment by Western blot analysis (Fig. S5 B). Relative to CHX treatment alone, inhibition of PDIA3 by pHMB prevented the accumulation of HC fragment, confirming that PDIA3 indeed cleaves antibody HC in the ER. Although PDIA3 inhibition did not block HC degradation, the clearance rate of HC was reduced 1.4-fold compared with CHX treatment alone, indicating that HC cleavage may accelerate HC degradation.

PDIA3 inhibition triggers a mild UPR phenotype and may lead to cell death. (A) RNAi-resistant PDIA3 WT or mutants were transiently expressed in mAb2-expressing cells. In cells cotransfected with PDIA3 siRNA, endogenous PDIA3 was depleted while the expression of the exogenous RNAi-resistant PDIA3 was not affected. (B) Cells expressing mAb2 HC were treated with CHX alone or in combination with PDIA3 inhibitor pHMB. Note that in CHX-treated samples, intracellular HC levels were reduced as HC fragment levels were increased, while in CHX- and pHMB-treated samples, intracellular HC fragment levels did not increase, and the levels of intact HC were relatively stable compared with CHX only–treated samples. The degradation rates of full-length HC in both conditions were calculated as the slope of the HC mass versus time graph. (C) Cells expressing mAb2 HC/LC were treated with indicated siRNA oligos and analyzed for levels of phosphorylated IRE1 and other indicated proteins. (D) Cells expressing mAb2 HC/LC were treated for 2 h with or without 1 mM DTT and analyzed for levels of phosphorylated IRE1. DTT treatment triggered UPR. (E) Two multiple myeloma cell lines (EJM and LP-1) were treated with PDIA3 inhibitor pHMB (250 µM) or vehicle for 24 h. Cells were then stained with annexin V-FITC and DNA-binding dye propidium iodide (PI) to indicate apoptotic and dead cells, respectively.
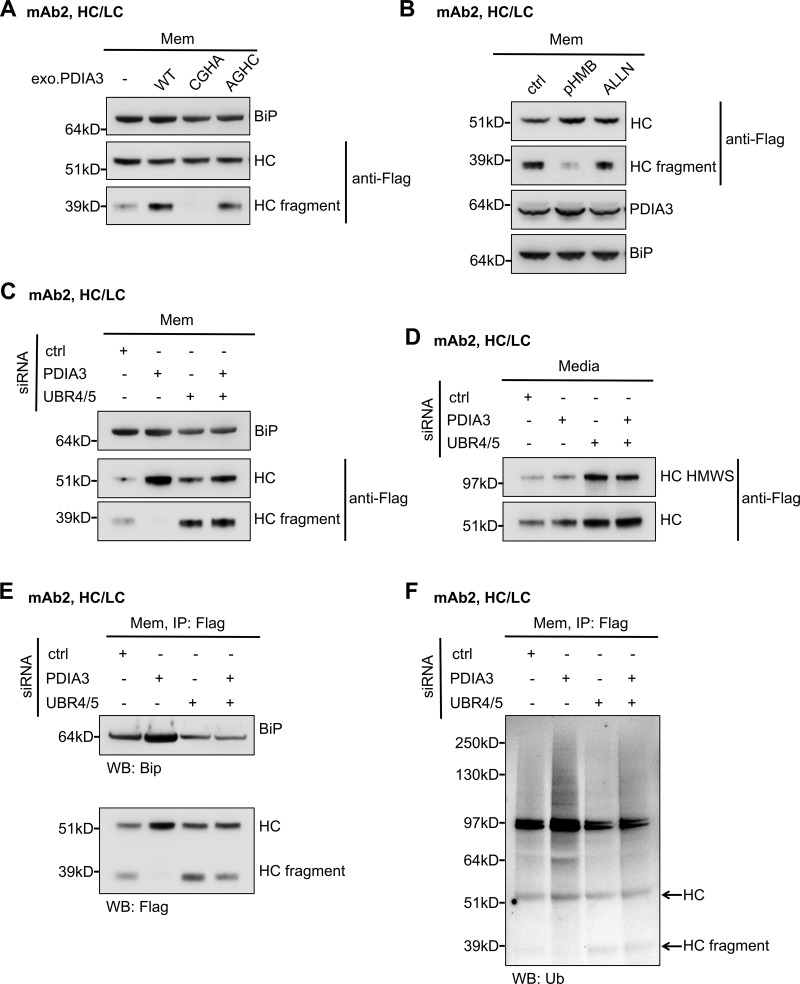
PDIA3 knockdown stabilizes ubiquitinated antibody HC. (A) Cells that express mAb2 HC/LC were transfected with PDIA3 WT or the indicated mutants and subjected to subcellular fractionation. HC and HC fragment associated with the membrane fraction were analyzed by Western blotting. CGHA: PDIA3 C60A/C409A mutant that has the second cysteine residues of the CGHC motifs mutated to alanine residues, abolishing protease activity. AGHC: C57A/C406A mutant with the first cysteine of CGHC motifs mutated to alanine, abolishing isomerase activity while maintaining partial protease activity. (B) Cells were treated with the indicated cysteine protease inhibitors for 16 h, and proteins associated with the membrane fraction were analyzed. pHMB (25 µM). ALLN (10 µM): N-acetyl-Leu-Leu-Norleu-al or Calpain Inhibitor I. (C–F) Cells were treated with the indicated siRNA oligos. HC species that associated with the membrane fraction (C) or media (D) were analyzed by Western blotting. HC immunoprecipitated from the membrane fractions were analyzed for HC levels and BiP association (E) as well as HC ubiquitination (F). Arrowheads indicate where HC and HC fragment should be localized.
To understand the function of PDIA3 in IgG HC degradation, we depleted PDIA3 alone, UBR4/UBR5, or all three (PDIA3, UBR4, and UBR5) in full mAb2-expressing cells. 3 d after siRNA transfection, the cells were pelleted, washed, and seeded into fresh media followed by 24 h of incubation to collect secreted HC (Fig. 4, C–F). Levels of UBR4, UBR5, and PDIA3 depletion by siRNA oligos were assessed (Fig. S5 C), and the intracellular membranes were isolated and analyzed (Fig. 4 C). Levels of antibody secretion in the last 24 h were also analyzed (Fig. 4 D). Additionally, we immunoprecipitated HC from the membrane fractions (Fig. 4 E) to determine its ubiquitination level (Fig. 4 F). In UBR4- and UBR5-depleted cells, the levels of both full-length HC and HC fragment were increased while the level of ubiquitinated HC was reduced compared with the control siRNA-treated cells (Fig. 4, C and F), as we previously observed (Fig. 2). The secretion of HC and HMWS of HC was also increased upon UBR4 and UBR5 depletion (Fig. 4 D). Together these results reaffirmed that UBR4/UBR5 are involved in HC and HC fragment ubiquitination and degradation. When PDIA3 alone was depleted, HC fragment was barely detectible, and the level of intracellular full-length HC significantly increased without a substantial increase in the levels of secreted HC (Fig. 4, C and D). This suggested that HC molecules might have been trapped in the ER. Furthermore, PDIA3 depletion increased levels of ubiquitinated HC (Fig. 4 F), implying that PDIA3-mediated HC cleavage played an important role for the extraction and degradation of ubiquitinated HC. We believe that without PDIA3 cleavage, the intact ubiquitinated HC molecules could be trapped in the translocon and not efficiently extracted for ERAD. Cleavage of HC by PDIA3 helps to accelerate the extraction of the N-terminal ubiquitinated HC fragments. Therefore, knocking down of PDIA3 stabilized ubiquitinated HC molecules, causing an increase in the levels of ubiquitinated HC. PDIA3 depletion also increased BiP (binding immunoglobulin protein) association with HC molecules (Fig. 4 E) and slightly elevated the level of phospho-IRE1 (Fig. S5, C and D), which are indicators of UPR and cellular stress (Frakes and Dillin, 2017). Interestingly, inhibition of PDIA3-mediated HC degradation in professional antibody-secreting cells, by PDIA3 inhibitor pHMB, induced cell death in several multiple myeloma cell lines (Fig. S5 E).
Depletion of PDIA3, UBR4, and UBR5 all together increased both intracellular and secreted HC levels, as observed for UBR4/UBR5 depletion, suggesting that UBR4 and UBR5 might function upstream of PDIA3. Presence of the intracellular HC fragment in PDIA3/UBR4/UBR5 triple-depleted cells, compared with the cells depleted for PDIA3 alone (Fig. 4 C), is likely due to diminished degradation of preexisting HC fragments. Likewise, the slight increase in the levels of ubiquitinated HC in triple-depleted cells compared with UBR4/UBR5 double-depleted cells is perhaps due to the stabilization of ubiquitinated HC molecules at the ER membrane due to PDIA3 depletion (Fig. 4 F). The intracellular HC levels in the UBR4/UBR5/PDIA3 triple knockdown cells was lower compared with PDIA3 single knockdown (Fig. 4 C), likely because in the absence of HC ubiquitination by UBR4 and UBR5, the unfolded HC molecules were no longer trapped in the translocon, and therefore could freely move back to ER lumen and be cleared by secretion into the medium (Fig. 4 D).
Based on these results, a possible mechanism of IgG HC degradation has been depicted in Fig. 5. (1) Unfolded IgG HC is recognized and brought to the ER/cytosol interface, where its N terminus is ubiquitinated by UBR4 and UBR5 in the cytosol. (2) As a multidomain protein, it may take lots of energy to unfold the entire HC molecule (Bhamidipati et al., 2005; Ruggiano et al., 2014) and a partially (CH2 and CH3 domains) folded HC molecule that is poly-ubiquitinated at the N terminus in the cytosol can neither be efficiently extracted from the ER nor be retrieved back into the ER lumen, thus clogging the translocon port. PDIA3 cleaves the partially translocated HC, accelerating the degradation of the poly-ubiquitinated HC N-terminal fragment and facilitating clearance of the clogged translocon (see Discussion below). (3) The remaining C-terminal HC fragment is then degraded by another round of recognition, ubiquitination, extraction, and degradation via the N-degron function of UBR4/UBR5. Depletion of UBR4/UBR5 did not abolish HC cleavage, suggesting that ubiquitination of HC is not a prerequisite to its cleavage as PDIA3 might cleave nonubiquitinated HC molecules inside the ER lumen. It is possible that other E3 ligases like Hrd1 are also involved in HC ubiquitination and degradation (Cattaneo et al., 2008). Interestingly, even in full mAb-expressing cells, there is still a portion of HC to be cleaved by PDIA3 and degraded by UBR4 and UBR5. It is possible that mainly the unassembled HC molecules are cleaved and degraded, while the HC molecules that assembled with LC are mostly secreted out of the cell.
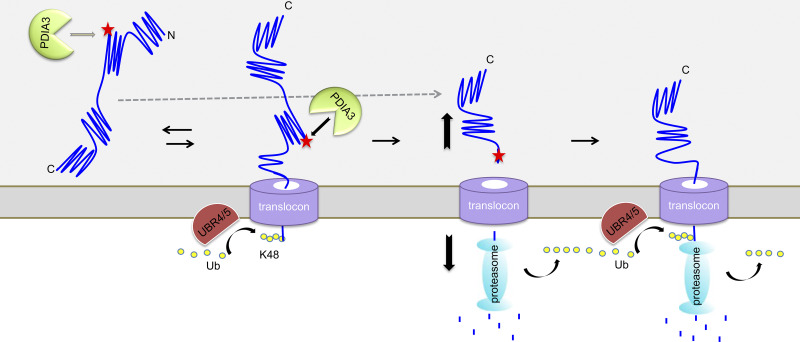
Model for UBR4/5-PDIA3 mediated coupled proteolysis/proteasome-dependent degradation of antibody HC. The N terminus of the unfolded antibody HC enters the translocon and is ubiquitinated by N-degrons UBR4 and UBR5. PDIA3 cleaves HC in the CH1 domain to allow fast degradation of the unfolded N-terminal half of HC and releases the C-terminal half from the translocon. The C-terminal half of the HC becomes now unfolded and a target for N-degrons. UBR4 and UBR5 further ubiquitinate and target the HC C-terminal fragment for degradation.
To assess fluctuation of UBR4, UBR5, and PDIA3 levels in IgG-expressing CHO cells, we compared the protein levels of UBR4, UBR5, and PDIA3 in seed train cells and cells in a fed batch antibody production culture. We analyzed two clones from different CHO hosts that express mAb4 (Fig. 6 A). We observed a drastic decrease in levels of UBR5 and a slight increase in levels of PDIA3 in production cultures compared with the seed train culture (Fig. 6 A). UBR4 levels were also slightly reduced during production, however not as drastically and consistently as UBR5 (data not shown). As expected, levels of HC and LC expression were higher in production culture (Fig. 6 A). This observation was further confirmed by analyzing UBR4, UBR5, and PDIA3 levels throughout a 14-d fed-batch antibody production process (data not shown). Additionally, decrease in UBR5 and to a lesser degree for UBR4 levels correlated to an increase in antibody productivity, as PDIA3 levels slightly increased later in the production run (Fig. 6 A and data not shown). These results suggest that during the fed-batch antibody production process, genes that are in involved in initiation of antibody ERAD might be down-regulated to circumvent ER and translocon congestion. On the other hand, PDIA3 up-regulation might alleviate blockage of ER translocons by cleaving away the folded segment of HC, allowing for faster degradation of HC fragment and clearing of ER translocons.
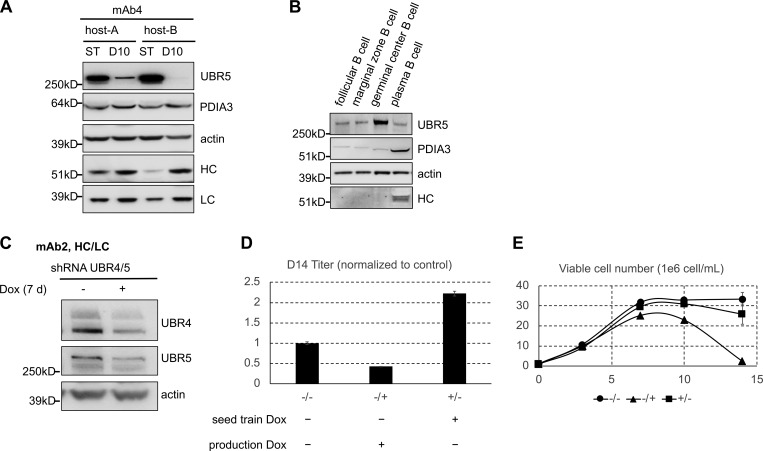
UBR5 level and antibody productivity. (A) Western blot analysis of seed train (ST) and day 10 (D10) production culture samples of mAb4 expressing clones for indicated proteins. (B) Intracellular levels of indicated proteins in different mouse B cell populations. (C) Dox induced knockdown of both UBR4 and UBR5 (UBR4/5) in pools of cells stably transfected with UBR4/5 shRNA construct. (D and E) Cells were treated with and without Dox for 4 d in the seed train, and these cultures were sourced to set up a 14-d fed-batch production assay with or without Dox. Day 14 (D14) titers (D) and viable cell densities on different days (E) during the production were measured.
Plasma B cells are the body’s natural antibody-expressing factories, and each of them is capable of secreting several thousand antibody molecules per second. They differentiate from B lymphocytes and entail the spectacular enlargement of ER to sustain massive Ig production (Calame et al., 2003). We assessed the protein levels of UBR5 and PDIA3 in mouse germinal center B cells, plasma cells, and several other types of B cells that do not produce IgG as controls. We observed that, similar to CHO cells, UBR5 level decreased while PDIA3 level increased in high antibody-producing plasma cells compared with low antibody-producing germinal center B cells (Fig. 6 B), suggesting that antibody degradation machinery and its reorganization might be conserved.
Since UBR4 and especially UBR5 were down-regulated in both CHO and plasma B cells, we decided to eliminate or down-regulate these proteins in CHO cells. UBR4 or UBR5 knockout hosts were generated; however, antibody-expressing cells derived from these hosts could not survive selection (data not shown), perhaps due to their inability to clear unfolded proteins and ER stress. Therefore, we generated a pool of cells that constitutively expressed mAb2 HC/LC and coexpressed UBR/UBR5 shRNAs under a tetracycline inducible Tet-On promoter. In the presence of doxycycline (Dox), knockdown efficiencies for UBR4 and UBR5 were ∼61% and 45%, respectively (Fig. 6 C). Continuous depletion of both UBR4 and UBR5 in the seed train gradually increased antibody secretion (data not shown). We then used untreated or UBR4/UBR5 knocked down (14 d) seed train cells to set up a fed-batch production assay (Fig. 6, D and E). We observed that UBR4/UBR5 knockdown during production affected cell growth and caused cultures to crash before day 10 (Fig. 6 E). Interestingly, knocking down UBR4/UBR5 in the seed train, before setting up the production, reduced cell death and led to an increase of 120% in titer (Fig. 6, D and E).
Altogether, these results suggest that down-regulation of UBR4/UBR5 may be a prerequisite for high antibody production, and knocking down UBR4 and UBR5 in the seed train may prompt the cells to switch from an antibody degradation mode to an antibody secretion mode. This could better prepare the cells for high antibody productivity once they were inoculated into the production culture.
A number of studies have identified key components of the ERAD pathway; however, the mechanism of ERAD is not fully understood. In yeast, two ERAD E3 ligases, Hrd1p and Doa10p, are found sufficient to recognize a diverse set of misfolded proteins (Mehnert et al., 2010), whereas in mammals, the repertoire of ERAD E3 ligases has been diversified (Bagola et al., 2011; Christianson et al., 2011; Olzmann et al., 2013; Smith et al., 2011; Vembar and Brodsky, 2008), presumably to cope with the higher degree of specialization and regulation required in complex multicellular organisms. Our study identified two novel ERAD-related E3 ligases, UBR4 and UBR5, that are also involved in the N-degron pathway (Dougan et al., 2012; Sriram et al., 2011). Although most substrates in the N-degron pathway discovered so far are cytosolic proteins, there are indications that N-recognins may also serve in the ERAD pathway. In yeast, Ubr1 is identified as an ERAD ubiquitin ligase and is involved in degradation of two transmembrane proteins (Stolz et al., 2013). Furthermore, it has been shown that ER chaperone BiP is degraded when its N-terminal amino acid is arginylated, generating another N-degron of the N-degron pathway (Shim et al., 2018). Therefore, it may be a common mechanism for N-recognins to participate in the ERAD of ER client proteins. Our data show that UBR4 and UBR5 are membrane-localized in antibody-expressing cells, suggesting they may have access to the ER substrates. After signal peptide cleavage, the first amino acid of our antibody HC is glutamine, which is a residue that can be arginylated (Arg) and then recognized by Arg/N-recognins including UBR4 and UBR5. Likewise, the PDIA3-cleaved C-terminal HC fragment starts with a threonine residue, which can be targeted for degradation following acetylation (Ac). Unlike the Arg/N-degron pathway, in mammalian cells the Ac/N pathway is poorly characterized, and Ac/N-recognins have not been identified (Gibbs et al., 2014; Sriram et al., 2011). Our result that the degradation of HC fragment also depends on UBR4 and UBR5 suggests that UBR4 and UBR5 may serve as Ac/N-recognins as well. Therefore, cleavage of HC into halves provides additional N-degrons, thus accelerating HC degradation.
In both the ERAD and N-degron pathways, multiple ubiquitin ligases can function together to target a protein for degradation (Ashton-Beaucage et al., 2016; Hwang et al., 2009, 2010; Morito et al., 2008; Younger et al., 2006). Similarly, we have found that both UBR4 and UBR5 are required for HC degradation, although they might also have overlapping functions. Whether these two E3 ligases form a complex or sequentially ubiquitinate HC requires further exploration. Last, UBR4 was previously suggested to mediate protein degradation by autophagy (Tasaki et al., 2013). However, for antibody HC degradation, we determined that the proteasome but not the lysosome is involved in HC degradation (Fig. 1 A), suggesting that UBR4 does not mediate autophagy-based degradation of the unfolded HC molecules. Besides UBR4 and UBR5, we have found that Hrd1 is also involved in HC degradation. Also, HC appears to have a very broad ubiquitination pattern involving different kinds of ubiquitin linkages, which suggests that multiple E3 ligases maybe involved in HC degradation. The initial N terminus ubiquitination of HC may be the first step to pull HC through the translocon to allow sequential ubiquitination of other lysine residues.
In this study, we have also identified PDIA3/ER-60 as the protease that mediates antibody HC quality control in the ER. In early studies of the ERAD pathway, there was significant interest in identifying an ER resident quality control protease. Many studies reported a requirement for proteolysis or a proteasome-independent ER-associated degradation pathway to remove unfolded ER resident or client proteins (Donoso et al., 2005; Loo and Clarke, 1998; Omura et al., 1992; Shenkman et al., 2007). The identification of PDIA3 made it a good candidate for the long-sought ER quality control protease (Adeli et al., 1997; Okudo et al., 2000; Otsu et al., 1995; Rutledge et al., 2013; Urade and Kito, 1992). For example, one ER substrate, ApoB100, is cleaved by PDIA3 to generate a 50-kD fragment (Qiu et al., 2004). However, there was no evidence for a broader role of PDIA3 in the degradation of other substrates (Lindquist et al., 1998; Needham and Brodsky, 2013; Oliver et al., 1999; Zhang et al., 2006). Our study identified IgG HC as a novel substrate of PDIA3, and also characterized a mechanism that couples it to the proteasomal degradation pathway. Besides PDIA3/ER-60, there are other proteases involved in ERAD that cleave unfolded proteins. A rhomboid family protein, RHBDL4, is an ER-resident intramembrane protease that interacts with and cleaves ubiquitinated type I single-spanning and polytopic membrane proteins with unstable transmembrane helices, allowing efficient turnover of the cleaved products (Fleig et al., 2012). Another intramembrane ER protease, SPP, cleaves type II transmembrane proteins with a short ER luminal tail. Substrate ubiquitination by E3 ligase TRC8 is required for SPP cleavage that is coupled to subsequent proteasomal degradation (Boname et al., 2014; Chen et al., 2014). Compared with transmembrane proteins, the dislocation of luminal ERAD substrates is more complex, as polypeptides have to be inserted into a translocon and moved across the ER membrane. A polypeptide chain may be able to slide back and forth across a translocon, but there has to be energy input to achieve net movement into the cytosol. Ubiquitination of the substrate may prevent it from sliding back into the ER lumen, and further extraction force provided by AAA-ATPase VCP/p97 or the AAA-ATPases in the 19S cap of the proteasome may pull the substrate out of the ER (Lee et al., 2004; Mayer et al., 1998; Oberdorf et al., 2006; Plemper et al., 1997; Ye et al., 2004). However, for multidomain proteins like antibody HC, it is possible that the unfolded part of the protein is inserted into the cytosol while the folded domains remain in the ER. It is known that the CH2 domain of the antibody HC folds independently, and disulfide bond formation facilitates this folding (Feige et al., 2004), while the folding of the CH1 domain is much more difficult, requiring the association with BiP and LC (Elkabetz et al., 2005; Feige et al., 2009; Marcinowski et al., 2011; Nishimiya, 2014). If the unfolded VH and CH1 domains of HC slide into the translocon and become ubiquitinated, while the CH2 and CH3 domains are still in their native form, the protein can neither slide back into the ER lumen nor be dislocated into the cytosol. This could clog the translocons. In this case, as we have shown in this study, PDIA3 can cleave the HC, allowing release of the folded C-terminal domain and degradation of the unfolded N-terminal domain, thereby clearing the translocon. A similar mechanism was characterized in translocation of newly synthesized proteins into the ER. For substrates whose translation and translocation are not tightly coupled, during their translocation they can undergo premature cytosolic folding, causing translocon clogging. In yeast and mammalian cells, respectively, the ER metalloprotease Ste24 and its human homologue ZMPSTE24 can approach the clogged translocon to cleave the clogged proteins and restore translocon function (Ast et al., 2016). Therefore, cleavage at the plane of the membrane seems to be a common mechanism to maintain ER homeostasis, as it reduces the energy required for dislocation and facilitates the extraction of the protein from the ER membrane.
Degradation of unfolded protein through the ubiquitin-proteasome system, however, is an energy-consuming process. This proteolysis/proteasomal coupled ERAD pathway might be sufficient to clear unfolded antibody from the ER when expressed at low to moderate levels as in recombinant CHO cells in the seed train culture or in nonplasma B cells. Once antibody expression maximizes, as in plasma B cells or CHO cells in production culture, increased ER throughput and accumulation of unfolded proteins might activate alternative pathways to clear the unfolded proteins. Consistent with this hypothesis, in plasma B cells and antibody-producing CHO cells (late in production culture) as cell division halts and the majority of cellular transcripts are that of the expressed antibody, the levels of UBR4 and UBR5 are down-regulated. Similarly, a previous study had shown a decrease of proteasome-associated proteins with increasing recombinant antibody-specific productivities (Sommeregger et al., 2016). Down-regulation of UBR4 and UBR5 could reduce ERAD-mediated degradation of antibody HC while concurrently increasing PDIA3 levels to help clear clogged translocons or aggregates of HC molecules in the ER lumen. Furthermore, in both plasma cells and CHO cells during antibody production, ER stress and the unfolded protein response are triggered (Cenci and Sitia, 2007; Ma and Hendershot, 2003). Therefore, it is plausible that large amounts of antibody expression could trigger a switch, diverting HC clearance from the ERAD pathway to that of secretion pathway. Consistent with this, knockdown of UBR4 and UBR5 in the seed train cells for a hard-to-express molecule mAb2 resulted in increase in antibody secretion once these cells were used to inoculate the production cultures (Fig. 6). A mAb molecule is often hard to express when the molecule has problems with folding and/or assembly (e.g., a molecule has large hydrophobic surface area). For cells expressing a hard-to-express molecule, it is possible that the knockdown of UBR4/UBR5 in the seed train mimics the reduced degradation effect caused by high antibody production and prompts the cells to switch to antibody secretion. It is also plausible that the knockdown of UBR4/UBR5 suppresses degradation of the unfolded HC molecules and increases their chances for folding and secretion. Therefore, knockdown of UBR4 and UBR5 may improve the productivity of the hard-to-express molecule.
In summary, we have identified a coupled-proteolysis/proteasome-mediated ERAD pathway involved in degradation of antibody HC. We have shown that the unfolded antibody HC interacts with and is ubiquitinated by E3 ligases in the N-degron pathway, UBR4 and UBR5, and is then extracted and degraded by the proteasome. ER luminal protease PDIA3 cleaves HC to accelerate HC degradation and clear the translocon. PDIA3-mediated cleavage also creates another N-terminal destabilizing residue, which initiates a further round of ubiquitination and degradation. We have also shown that the ERAD-mediated degradation of antibody HC is down-regulated once antibody production by cells is ramped up, suggesting that UBR4 and UBR5 may be suitable engineering targets to increase CHO cell protein productivity. We plan to further pursue the role of these proteins in antibody-expressing B cells.
CHO cells were cultured in a proprietary DMEM/F12-based medium in 125-ml shake flask vessels in an incubator with 150-rpm shaking speed, 37°C, and 5% CO2. Cells were passaged at a seeding density of 4 × 105 cells/ml every 3–4 d. For mAb, HA-Ub, or shRNA stable transfection, CHO cells were transfected using the MaxCyte STX Transfection System according to the manufacturer’s recommendation (MaxCyte). Transfected cells were single-cell plated into 384-well plates filled with DMEM/F12-based selective medium. After 3 wk, clones from wells with >25% confluence were selected and expanded. Cell lines that expressed transfected gene with acceptable levels, as examined by Western blotting, were used for further experiments. For pool selection, transfected cells were seeded into selective medium and cultured in shake flasks. The viabilities and viable cell counts for the selected pools were analyzed every 3–4 d. Once the viability exceeded 90%, the pools were used for further experiments.
Fed-batch production cultures were performed in shake flasks with proprietary chemically defined basal medium along with bolus proprietary feeds on days 3, 7, and 10. A temperature shift from 37°C to 35°C was performed on day 3. Day 7, 10, and 14 titers were determined using protein-A affinity chromatography with UV detection. Percent viability and viable cell counts were determined using a Vi-Cell XR instrument (Beckman Coulter).
For mAb1, mAb2, and mAb3 expression, IgG HC with a C-terminal Flag sequence was cloned into a mammalian expression vector alone or together with LC with a C-terminal HA sequence. For ubiquitin expression, an N-terminal HA tag was linked to WT or mutated ubiquitin by PCR. The HA-Ub fragment was then cloned into pcDNA3.1(-) using EcoRI and BamHI sites. Partial or full cDNA sequences of UBR4, UBR5, PDIA3, and other proteases in our CHO K1 host were determined by PCR, and siRNA sequences were then designed based on them using the online software Designer of Small Interfering RNA. A nonspecific siRNA was used as negative control. For siRNA transfection, 100 nM for each siRNA oligo was used. The top two siRNA sequences for each gene, tested by transient transfection, were converted to shRNA and cloned into a Tet-On inducible shRNA expression vector. For UBR4 and UBR5 knockout, gRNAs targeting to both the 5′ and the 3′ ends of the genes were cloned into a gRNA-expression vector. This construct was then cotransfected with Cas9 expression plasmid into CHO-K1 host cells. Transfected cells were single-cell cloned, and the knockouts of UBR4 or UBR5 for selected clones were analyzed by Western blotting: control siRNA (sense): 5′-GAAUUAUAAUGCUUAUCUA-3′; UBR4-siRNA-1 (antisense): 5′-UGAGUGAUGAAACUACAGCUG-3′; UBR4-siRNA-2 (antisense): 5′-UACAUUAGUGACAUAGAAGGG-3′; UBR5-siRNA-1 (antisense): 5′-UAGAAAGUGAUGAACGACGAA-3′; UBR5-siRNA-2 (antisense): 5′-UGUGAAAUCAGCUCCUCGGGG-3′; PDIA3-siRNA-1 (antisense): 5′-UUACAGUGACCACACCAAGGG-3′; PDIA3-siRNA-2 (antisense): 5′-AUCAUCAUACUCCUUCACCAG-3′; Hrd1-siRNA-1 (antisense): 5′-UAUGUUGCGCAGACUCUGCAG-3′; Hrd1-siRNA-2 (antisense): 5′-UGAGGUAGUAGGCAUGAGCCA-3′; UBR4-5′-gRNA targeting sequence: 5′-TAAGGCCTCAGCGGGTAACAGGG-3′; UBR4-3′-gRNA targeting sequence: 5′-GCTCTAGAGAGCCTAGGAGGAGG-3′; UBR5-5′-gRNA targeting sequence: 5′-GGCTGTTGAACTGTTCCGAGGG-3′; and UBR5-3′-gRNA targeting sequence: 5′-AGAATCTTCACTCGCTAGGAGG-3′.
The following antibodies were used: goat anti-human IgG-HRP (MP Biomedicals, 0855252), rabbit anti-BiP (C50B12, Cell Signaling Technology, 3177), rabbit anti-calnexin (Enzo Life Sciences, ADI-SPA-860), rabbit anti-UBR4 (Bethyl Laboratories Inc., A302-278A), rabbit anti-UBR5 (Bethyl Laboratories Inc., A300-573A), rabbit anti-Hrd1 (Abcepta Inc., AP2184A) rabbit anti-PDIA3 (Bethyl Laboratories, Inc., A305-258A), rabbit anti-phosphor-IRE1 (in-house raised), rabbit anti-IRE1 (in-house raised), mouse anti-β-actin-HRP (AC-15) (Abcam, ab49900), mouse anti-Flag-HRP (M2) (Sigma-Aldrich, A8592), mouse anti-HA-HRP (3F10) (Roche, 12158167001), mouse anti-ubiquitin-HRP (P4D1) (Santa Cruz Biotechnology, sc-8017), goat anti–mouse HRP (Santa Cruz Biotechnology, sc-2005), and donkey anti–rabbit HRP (Jackson ImmunoResearch Laboratories, Inc., 711–035-152).
To separate intracellular membranes from cytosol, 10 million cells were first washed with cold PBS and resuspended into 100 µl low-salt homogenization buffer (10 mM Tris, pH 7.5, 10 mM KCl, 2 mM MgCl2, and 2 mM DTT, with protease inhibitors cocktail) and kept on ice for 20 min. Protease inhibitor cocktail tablets were purchased from Roche (04693159001). Cells were then homogenized by passing through a 26G × 3/8 (0.45 mm × 10 mm) needle (BD Biosciences) 40 times (each time that cells were aspirated into and ejected from the syringe was counted separately). 1 μl of the cell homogenate was stained by trypan blue solution to examine the percentage of cell lysis by white-light microscopy. Normally, after homogenization, 10–20% of the cells remained intact. The homogenate was then spun at 1,000 g at 4°C for 10 min to pellet the unbroken cells, nuclei, and plasma membranes. The supernatant was then transferred to a 1-ml Beckman centrifuge tube (11 × 34 mm, Beckman Coulter Life Sciences, 343778) for ultracentrifugation at 100,000 rpm (539,994 g) for 30 min using TLA-120.2 rotor (Beckman Coulter Life Sciences) and Optima MAX-XP ultracentrifuge (Beckman Coulter Life Sciences). After centrifugation, the supernatant was transferred to a 1.5-ml tube as the cytosol fraction. The pellet was solubilized in 100 µl lysis buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5% NP-40, and 2 mM DTT, with protease inhibitors cocktail) as the intracellular membrane fraction. To determine the subcellular localization of HC or UBR4 and UBR5, equal volumes of cytosol or solubilized membranes were loaded on the same gel for comparison.
To exclude the possibility that UBR4 and UBR5 might be localized to other membrane organelles than ER, the pellets generated from 1,000-g centrifugation or 100,000-rpm ultracentrifugation in the previous step were further fractionated using the Thermo Scientific Subcellular Protein Fractionation Kit for Cultured Cells. Proteins extracted from different cellular compartments at each step were analyzed by SDS-PAGE and Western blot.
For density gradient, the 10–30% OptiPrep (iodixanol) gradient was prepared as described (Li and Donowitz, 2014). The total membranes and cytosol from the cells that were prepared by homogenization and brief centrifugation were loaded on the top of the gradient. The gradient was then centrifuged 100,000 g for 16 h in a SW40 swinging bucket (Beckman Coulter Life Sciences). 13 fractions (1 ml each fraction) were then collected from the top to the bottom of the gradient. The same volume of each fraction was analyzed by SDS-PAGE and Western blot.
Cells were washed with PBS and resuspended in fresh media containing 20 µg/ml CHX. In some cases, 25 µM pHMB was also added into the cell culture to prevent HC cleavage. At each time point, three million cells were collected from the cell culture and pelleted. Each cell pellet was lysed in 50 µl lysis buffer. An equal amount of total protein for each lysate sample was used for SDS-PAGE and Western blot analysis.
For immunoprecipitation, protein concentration of total cell lysate, cytosol, or solubilized membrane fraction was measured by BCA assay (Thermo Fisher Scientific). An equal amount of total protein from each sample was used for immunoprecipitation. IgG HC was precipitated either by protein A/G agarose (Thermo Fisher Scientific) or by anti-Flag M2 agarose (Sigma-Aldrich). Cell lysate, cytosol, solubilized membrane fraction, or cell culture (harvest cell culture fluid [HCCF]) was incubated with protein A/G agarose or anti-Flag M2 agarose for 2 h at 4°C with rotation. Beads were then pelleted, washed twice by cold lysis buffer or PBS, and boiled in SDS sample buffer.
Cell lysate, cell culture HCCF, cytosol, solubilized membrane fraction, and immunoprecipitated samples as described above were analyzed by Western blotting. To compare protein levels in cells with different treatment, an equal amount of cell lysate or solubilized membranes for each sample was loaded onto the same SDS-PAGE gel. Actin, BiP, or calnexin was probed as loading control. For HCCF and immunoprecipitated samples, the same number of cells was used in the experiment, and the same volume of each sample was analyzed by SDS-PAGE.
Bands of interest on SDS-PAGE gel were excised, washed, and destained in 50 mM ammonium bicarbonate in 50/50 acetonitrile/water (vol/vol; 100 µl, 20 min). Gel piece(s) were dehydrated with acetonitrile and digested with 0.2 µg trypsin (Promega), in ammonium bicarbonate, pH 8.0, overnight at 37°C. Peptides were extracted from the gel slice(s) in 50 µl of 50/50 acetonitrile/1% formic acid (vol/vol; Sigma-Aldrich) for 30 min followed by 50 µl of pure acetonitrile. Extractions were pooled and evaporated to near dryness and were reconstituted in 2% acetonitrile/0.1% formic acid (vol/vol). Samples were injected via an auto-sampler onto a 75 µm × 100 mm column (BEH [ethylene bridged hybrid], 1.7 micron, Waters Corp.) at a flow rate of 1 µl/min using a NanoAcquity UPLC (Waters Corp). A 40-min gradient from 98% solvent A (water + 0.1% formic acid) to 80% solvent B (acetonitrile + 0.08% formic acid) was used to separate the samples. Samples were analyzed in-line via nanospray ionization into a hybrid LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). Data were collected in data-dependent mode with the parent ion being analyzed in the Orbitrap and the top eight most abundant ions being selected for fragmentation and analysis in the ion trap. Tandem mass spectrometric data were analyzed using the Mascot search algorithm (Matrix Sciences). For N-terminal sequencing, Edman degradation was performed as previously reported (Henzel et al., 1987, 1999).
For ubiquitin linkage analysis, six regions above the 55-kD HC band to the top of the gel at the sample input well were excised. Excised gel bands were destained, dehydrated, and trypsin-digested as described above. Peptides were extracted with 50% acetonitrile/0.1% trifluoroacetic acid followed by 100% acetonitrile. At this point, the six individual digests were combined, and an isotopically labeled synthetic peptide (Cell Signaling Technology) mixture containing peptides representing ubiquitin chain linkages as they would be observed after trypsin digestion (i.e., Gly-Gly additions to specific lysines representing poly-ubiquitin chains) was spiked into the extraction at a known concentration and then dried. Ubiquitin peptides included in the experiment were described previously (Phu et al., 2011). Peptides were reconstituted in 2% acetonitrile/0.1% formic acid to a final spiked peptide concentration of 100 femto-mole/µl and analyzed by mass spectrometry. Peptides were separated using a Thermo UltiMate 3000 RSLCnano Proflow system (Thermo Fisher Scientific) with a gradient of 2% buffer A (98% H20, 2% acetonitrile with 0.1% formic acid) to 30% or 40% buffer B (98% acetonitrile, 2% H20, 0.1% formic acid) with a flow rate of 300 or 350 nl/min. Samples were separated over a 25-cm IonOpticks Aurora Series column (IonOpticks). All samples were analyzed within a 60-min nLC-MS/MS analysis using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Precursor masses of heavy and light peptides were extracted to determine the peak area under the curve for quantitation. Peptide extraction criteria were as follows: ±10 ppm mass tolerance and ±0.5 min retention time across all time points. All peak integration was manually verified and area under the curve calculated for total content of specific peptides.
WT mice with C57/BL6 background were purchased from Jackson Laboratories. All mice were maintained under specific pathogen-free conditions. All animal experimentation protocols were approved by the Laboratory Animal Resources Committee at Genentech Inc.
C57/BL6 mice were immunized via intraperotoneal injection with 100 µl of emulsion containing 50 µg of KLH (keyhole limpet haemocyanin) in 50 µl PBS and 50 µl complete Freund’s adjuvant on days 0, 14, and 28. Spleens from the immunized mice were collected 14 d after last immunization. Total spleen cells were stained with fixable viability dye (eBioscience) in PBS for 10 min at room temperature, followed by one wash with cold PBS/0.2% BSA. To avoid nonspecific binding, cells were blocked with TruStain fcXTM (Biolegend) in PBS/0.2% BSA at 4°C. Antibodies against surface antigens were added 15 min after Fc block without washing, for 30 min at 4°C, followed by three washes with 1 ml cold PBS. Follicular B cells (CD19+IgM+IgD+CD21−CD23+), marginal zone B cells (CD19+IgD+IgMhighIgDlowCD21+CD23−), germinal center B cells (CD19+IgD-CD95+GL-7+), and plasma cells (CD19midCD138+) were sorted using a BD FACSAria III following standard procedures. Sorted cells were lysed by radioimmunoprecipitation assay buffer in the presence of 1× protease inhibitor (Thermo Fisher Scientific) immediately after FACS sorting. Protein expression in each subset was examined using Western blot analysis. The following antibodies were used to label splenocytes: CD19–Alexa Fluor 700 (clone 6D5), CD138-BV605 (clone 281–2), GL-7-PE (clone GL7), IgD-PE/Cy7 (clone 11-26c.2a), IgM-FITC (clone RMM-1), and CD95-APC (clone SA367H8; Biolegend). All antibodies were used at 1 µg/ml. Fc block was used at 2.5 µg/ml. Fixable Viability Dye eFluor 780 was used at a 1:1,000 dilution to gate out dead cells.
Multiple myeloma cell lines EJM and LP-1 were thawed and cultured in RPMI 1640/10% FBS/1 × glutamine media. Cells were treated with 250 µM pHMB or vehicle for 24 h and analyzed for cell death and/or apoptosis by staining using the Dead Cell Apoptosis Kit with Annexin V FITC and propidium iodide (Thermo Fisher Scientific, V13242) followed by FACS analysis.
Genomic DNA and RNA samples were purified using Qiagen AllPrep DNA/RNA Mini Kit (80204) according to the manufacturer’s instructions. To determine both gene copy number and mRNA level of HC, TaqMan RT-PCR assays were performed using TaqMan Universal Master Mix (Thermo Fisher Scientific, 4304437). RNA was first reverse-transcribed into cDNA using SuperScript II reverse transcription kit (Thermo Fisher Scientific, 18064014). Each TaqMan RT-PCR reaction contained 1× PCR Master mix, 500 nM forward primer, 500 nM reverse primer, 200 µM probe, and purified genomic DNA or cDNA sample. The thermal cycling conditions were 30 min at 48°C and 10 min at 95°C, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All reactions were processed on the 7900 HT Fast Real Time PCR System (Life Technologies). Data were analyzed using SDS v2.4 software (Life Technologies) after TaqMan RT-PCR amplification. The HC gene copy numbers were normalized based on the defined copy number of reference genes Bax, albumin, Hprt, and β-microglobulin. The relative expression levels of HC were determined based on the Delta Ct analysis method. The expression level of reference gene, β-microglobulin, was used to normalize different RNA samples in each reaction. The primers used in this study were designed using primer express v3.0 (Life Technologies). The sequences of primers are as follows: HC, forward primer, 5′-TCAAGGACTACTTCCCCGAACC-3′; reverse primer, 5′-TAGAGTCCTGAGGACTGTAGGACAGC-3′; probe, 5′-FAM-ACGGTGTCGTGGAACTCAGGCGC-TAMRA-3′; LC, forward primer, 5′-TGACGCTGAGCAAAGCAGAC-3′; reverse primer, 5′-CAGGCCCTGATGGGTGAC-3′; probe, 5′-FAM-ACGAGAAACACAAAGTCTACGCCTGCGA-TAMRA-3′; Bax, forward primer, 5′-ACACTGGACTTCCTCCGAGA-3′; reverse primer, 5′-GCATTAGGAAGTTTGAGAACCA-3′; probe, 5′-FAM-CCCAGCCACCCTGGTCTTGG-TAMRA-3′; albumin, forward primer, 5′-TTCGTGACAGCTATGGTGAACTG-3′; reverse primer, 5′-GGTCATCCTTGTGTTTCAGGAAA-3′; probe, 5′-FAM-CTGTGCAAAACAAGAACCCGAAAGAAACC-TAMRA-3′; Hprt, forward primer, 5′-AAAGGACATAATTGACACTGGTAAA-3′; reverse primer, 5′-CAATTGCTTATTGCTCCCAA-3′; probe, 5′-FAM-CTGCTTTCCCTGGTCAAGCGG-TAMRA-3′; β-microglobulin, forward primer, 5′-CTTCCTGAACTGCTATGTGTCTCAA-3′; reverse primer, 5′-CCATCTTCTTTCCATTCTTCAACA-3′; and probe, 5′-VIC-TTCATCCCCCCCAAATTGAAATCGA-BHQ1-3′.
Fig. S1 shows the cell lines used in the co-immunoprecipitation study. Fig. S1 is linked to Fig. 1 D. Fig. S2 shows the subcellular localization of UBR4 and UBR5. Fig. S2 is linked to Fig. 1 B. Fig. S3 shows that IgG HC associates with UBR4 and UBR5, HC cleavage and ubiquitination, and knockdown of Hrd1. Fig. S3 is linked to Fig. 1 and Fig. 2. Fig. S4 shows that UBR4 and UBR5 are involved in HC degradation in multiple different cell lines. Fig. S4 is linked to Fig. 2. Fig. S5 shows the activity of PDIA3 in HC cleavage. Fig. S5 is linked to Fig. 3 and Fig. 4.
We would like to thank the Analytical Operations groups and their respective team members in Genentech who performed the standard analytical work. We would also like to thank Dr. Steven Lang and Dr. David Shaw for their critical review of the manuscript. We thank Dr. Nobuhiko Kayagaki and Dr. Vishva Dixit (Genentech Inc., South San Francisco, CA) for chain-specific K48, K63, and K11 mutant ubiquitin vector constructs.
The authors declare no competing financial interests.
Author contributions: D. Tang designed and performed the experiments, D. Tang, C. Lam, S. Louie, and D. Roy generated the cell lines that were used in this study, B. Haley designed shRNA constructs, C. Lam, P. Liu, and W. Sandoval performed the mass spectrometry analysis, D. Xue contributed to B cell subtype isolation, T. Patapoff contributed to protein model simulation, S. Misaghi and B. Snedecor supervised the project, and D. Tang and S. Misaghi wrote the article in consultation with B. Snedecor.
 UBR E3 ligases and the PDIA3 protease control degradation of unfolded antibody heavy chain by ERAD
UBR E3 ligases and the PDIA3 protease control degradation of unfolded antibody heavy chain by ERAD
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

