 Site-Selective
Csp3–Csp/Csp3–Csp2 Cross-Coupling Reactions
Using Frustrated Lewis Pairs
Site-Selective
Csp3–Csp/Csp3–Csp2 Cross-Coupling Reactions
Using Frustrated Lewis Pairs
Journal of the American Chemical Society


- Altmetric
The donor–acceptor ability of frustrated Lewis pairs (FLPs) has led to widespread applications in organic synthesis. Single electron transfer from a donor Lewis base to an acceptor Lewis acid can generate a frustrated radical pair (FRP) depending on the substrate and energy required (thermal or photochemical) to promote an FLP into an FRP system. Herein, we report the Csp3–Csp cross-coupling reaction of aryl esters with terminal alkynes using the B(C6F5)3/Mes3P FLP. Significantly, when the 1-ethynyl-4-vinylbenzene substrate was employed, the exclusive formation of Csp3–Csp cross-coupled products was observed. However, when 1-ethynyl-2-vinylbenzene was employed, solvent-dependent site-selective Csp3–Csp or Csp3–Csp2 cross-coupling resulted. The nature of these reaction pathways and their selectivity has been investigated by extensive electron paramagnetic resonance (EPR) studies, kinetic studies, and density functional theory (DFT) calculations both to elucidate the mechanism of these coupling reactions and to explain the solvent-dependent site selectivity.
Introduction
Frustrated Lewis pairs (FLPs) have garnered much attention over the last two decades, with numerous FLP systems being reported in the literature.1 The cooperative reactivity of the Lewis acidic and basic components has led to a plethora of different small-molecule activation reactions2 and catalysis.3 Current studies have focused on using FLP systems as alternatives or complementary systems to the use of transition-metal catalysts in organic synthesis.4 Recently, new reactivities of FLPs have been disclosed, indicating that Lewis acids and bases undergo single electron transfer (SET) events5 depending on the energy required (thermal or photochemical) to promote the FLP into a frustrated radical pair (FRP) system. In these instances, an electron is transferred from the donor Lewis base (LB) to the acceptor Lewis acid (LA) to generate a reactive FRP. Indeed, we and others have postulated that such radical reactivity may be taking place in the reactions of the B(C6F5)3/Mes3P FLP with certain substrates.6 The radical reactivity of FLPs has the potential to open up new opportunities for metal-free synthesis. In a previous study of the B(C6F5)3/Mes3P FLP with diaryl esters and alkenes, we observed Csp3–Csp2 coupling reactions. We proposed a radical mechanism for the reaction based on the observation of [Ar2CH]• and [Mes3P]•+ in electron paramagnetic resonance (EPR) studies (Scheme 1A).

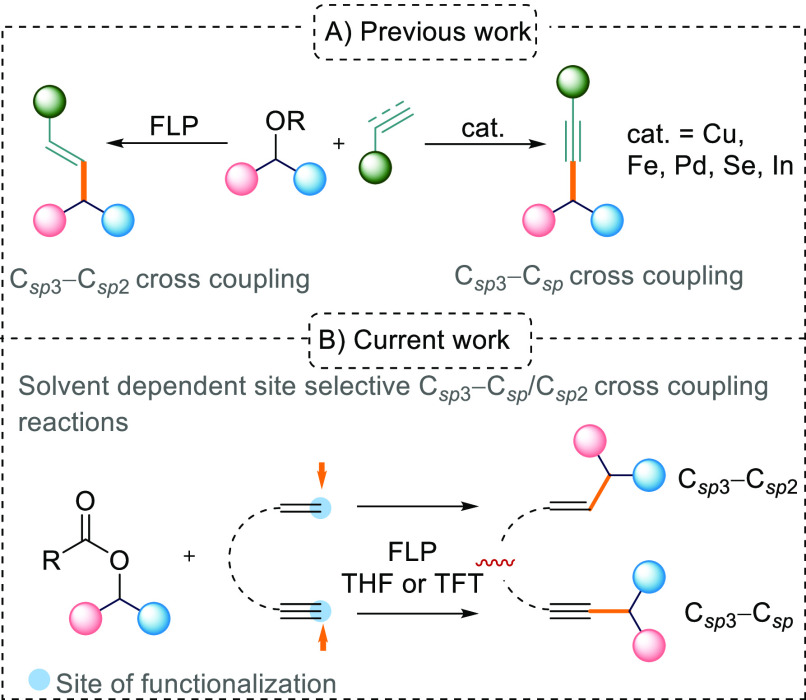
(A) Previous Work on Metal-Catalyzed Csp3–Csp Cross-Coupling Reactions and FLP-Mediated Csp3–Csp2 Cross Coupling and (B) This Work on FLP-Mediated, Solvent-Dependent, Site-Selective Csp3–Csp/Csp3–Csp2 Cross-Coupling Reactions
In this current study, we were interested in the reactions of FLPs with alkynes in the presence of aryl esters (Scheme 1B). The 1,2-trans-addition of the Lewis acidic and basic components of FLPs to alkynes is well established7 and has also been employed in catalytic transformations.8 Interestingly, when using terminal acetylenes such as phenylacetylene (PhC≡CH) with stronger bases such as tBu3P or TMP, deprotonation occurs instead of 1,2-addition to the alkyne generating [LBH][PhC≡CB(C6F5)3] salts.7c In transition-metal chemistry, the activation of terminal alkynes for cross-coupling reactions is commonplace in the synthetic chemist’s toolbox to construct carbon–carbon bonds.9 For example, palladium- or copper-catalyzed Sonogashira cross-coupling reactions of terminal alkynes with aryl or alkenyl halides have been used for Csp3–Csp2 coupling.10 In this study, we are interested in the less well reported Csp3–Csp coupling reactions. Typically, palladium11 or earth-abundant metal catalysts such as iron12 and copper13 are employed for these reactions, although examples are known with other elements such as indium14 as well as stoichiometric reactions using Brønsted15 and Lewis acids.16
Herein, we report the high reactivity of frustrated Lewis pairs in selective Csp3–Csp coupling reactions between aryl esters and terminal alkynes or 1-ethynyl-4-vinylbenzene. We also report solvent-dependent site selectivity when using 1-ethynyl-2-vinylbenzene as a substrate leading to selective Csp3–Csp or Csp3–Csp2 cross-coupling depending upon the solvent employed.
Results and Discussion
Reaction Optimization and Development
Initially, the FLP-mediated Csp3–Csp cross-coupling reaction between bis(4-fluorophenyl)methyl-4-fluorobenzoate (1a) and phenylacetylene was investigated using a range of reaction conditions (Table 1). As expected, no reaction occurred in the absence of an FLP (Table 1, entry 1). Reaction with only the Lewis base component of the FLP (Mes3P) showed no cross-coupled product, and a stoichiometric or catalytic (10 mol %) amount of the Lewis acid B(C6F5)3 led to only 22 and 5% isolated yields of the desired Csp3–Csp cross-coupled product, 2a (Table 1, entries 3 and 4). Stoichiometric amounts of both a Lewis acid and Lewis base were required for the Csp3–Csp coupling reaction to attain satisfactory yields of the cross-coupled products. The B(C6F5)3/Mes3P FLP in toluene at 70 °C led to the Csp3–Csp cross-coupled product, 2a, being formed in 54% yield (Table 1, entry 5). The optimum temperature was 70 °C with both lower (21 °C) and higher (110 °C) temperatures giving reduced yields (18 and 45% respectively) (Table 1, entries 6 and 7). More polar THF gave the highest yield of 83%, with trifluorotoluene (TFT) giving a 71% yield. CH2Cl2 and hexane, on the other hand, showed poorer or low yields (Table 1, entries 8–11). Interestingly, other Lewis acid boranes (BF3·OEt2 and BPh3) as well as Brønsted acids (CF3SO3H) showed no product formation when combined with Mes3P (Table 1, entries 12–14). Other basic phosphines including tBu3P, Ph3P and o-tol3P had complicated reaction mixtures with no or moderate yields of 2a being formed (Table 1, entries 15–17). Nitrogen Lewis bases including TMP (2,2,6,6-tetramethylpiperidine), DABCO (1,4-diazabicyclo(2,2,2)octane), and DMA (4-bromo dimethyl aniline) led to no product formation (Table 1, entries 18–20). The optimum conditions for the reaction were therefore chosen to be the use of the B(C6F5)3/Mes3P FLP in THF at 70 °C for 22–24 h.

| entry | LA | LB | solvent | temp (°C) | yield (%) |
|---|---|---|---|---|---|
| 1 | toluene | 70 | 0 | ||
| 2 | Mes3P | toluene | 70 | 0 | |
| 3 | B(C6F5)3 | toluene | 70 | 22 | |
| 4b | B(C6F5)3 | toluene | 70 | 5 | |
| 5 | B(C6F5)3 | Mes3P | toluene | 70 | 54 |
| 6 | B(C6F5)3 | Mes3P | toluene | 21 | 18 |
| 7 | B(C6F5)3 | Mes3P | toluene | 110 | 45 |
| 8 | B(C6F5)3 | Mes3P | THF | 70 | 83 |
| 9 | B(C6F5)3 | Mes3P | TFT | 70 | 71 |
| 10 | B(C6F5)3 | Mes3P | CH2Cl2 | 45 | 38 |
| 11 | B(C6F5)3 | Mes3P | hexane | 70 | 0 |
| 12 | BF3·OEt | Mes3P | toluene | 70 | 0 |
| 13 | BPh3 | Mes3P | toluene | 70 | 0 |
| 14 | CF3SO3H | Mes3P | THF | 70 | 0 |
| 15 | B(C6F5)3 | tBu3P | toluene | 70 | 45 |
| 16 | B(C6F5)3 | Ph3P | THF | 70 | 0 |
| 17 | B(C6F5)3 | o-tol3P | THF | 70 | 0 |
| 18 | B(C6F5)3 | TMP | THF | 70 | 0 |
| 19 | B(C6F5)3 | DABCO | THF | 70 | 0 |
| 20 | B(C6F5)3 | DMA | THF | 70 | 0 |
a All of the reactions were carried out on a 0.1 mmol scale, and reported yields are isolated. All reactions were carried out for 20–22 h.
b 10 mol % B(C6F5)3.
Reaction Scope
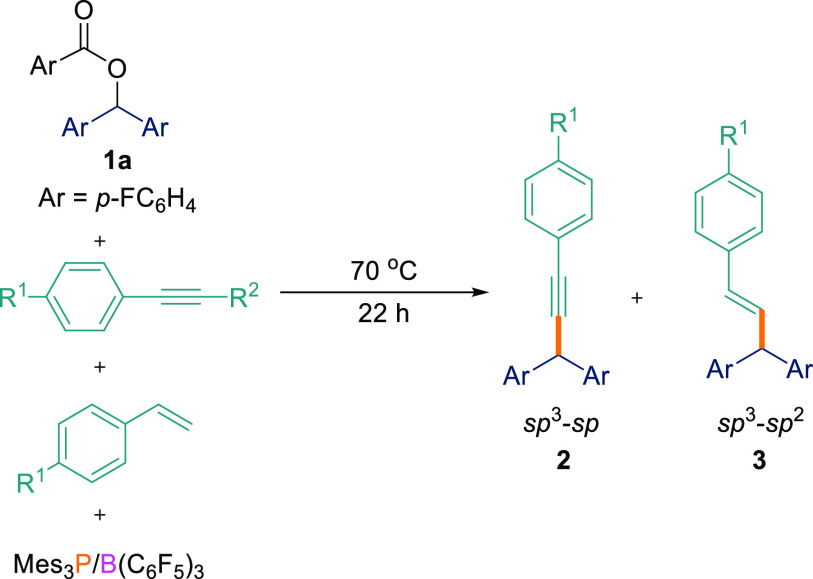
With the optimized reaction conditions in hand, several aryl esters (1a–l) were tested for the FLP-mediated Csp3–Csp coupling reaction with various acetylenic compounds (Scheme 2). Diaryl esters bearing electron-withdrawing/π-releasing (p-F, 1a; p-Cl, 1b), neutral (p-H, 1c), and electron-donating (p-OMe, 1d) groups all worked well for the reactions when coupled with aryl-substituted terminal acetylenes with electron-releasing (p-OMe, p-tBu), neutral (p-H), electron-withdrawing/π-releasing (p-F, p-Cl), and electron-withdrawing (p-CF3) groups generating products 2a–q in 60–85% yields. Asymmetrical diaryl esters 1e and 1f were also found to undergo the Csp3–Csp cross-coupling reaction, with several alkynes generating C–H-functionalized products 2r–w in excellent isolated yields (69–80%). We could also use alkyl/aryl esters containing just one aryl-stabilizing group. 1g could be cross-coupled with electron-neutral (phenylacetylene) and electron-releasing (4-ethynylanisole) alkynes to give 2x and 2y albeit in slightly lower yields of 61 and 65%, respectively. However, when cyclohexyl(phenyl)methyl-4-fluorobenzoate (1h) was employed, poor conversion was observed. Diaryl esters bearing strongly electron-withdrawing (p-CF3, 1j) groups also failed to react at all with terminal alkynes. We attribute this to the electron-deficient nature of the ester which is not Lewis basic enough to form an adduct with the Lewis acidic borane in the initial step of the reaction as observed by in situ11B and 1H NMR spectroscopy. The unwillingness of ester 1j to react with arylacetylenes can also be interpreted from DFT calculations (see later and SIFigure S180).
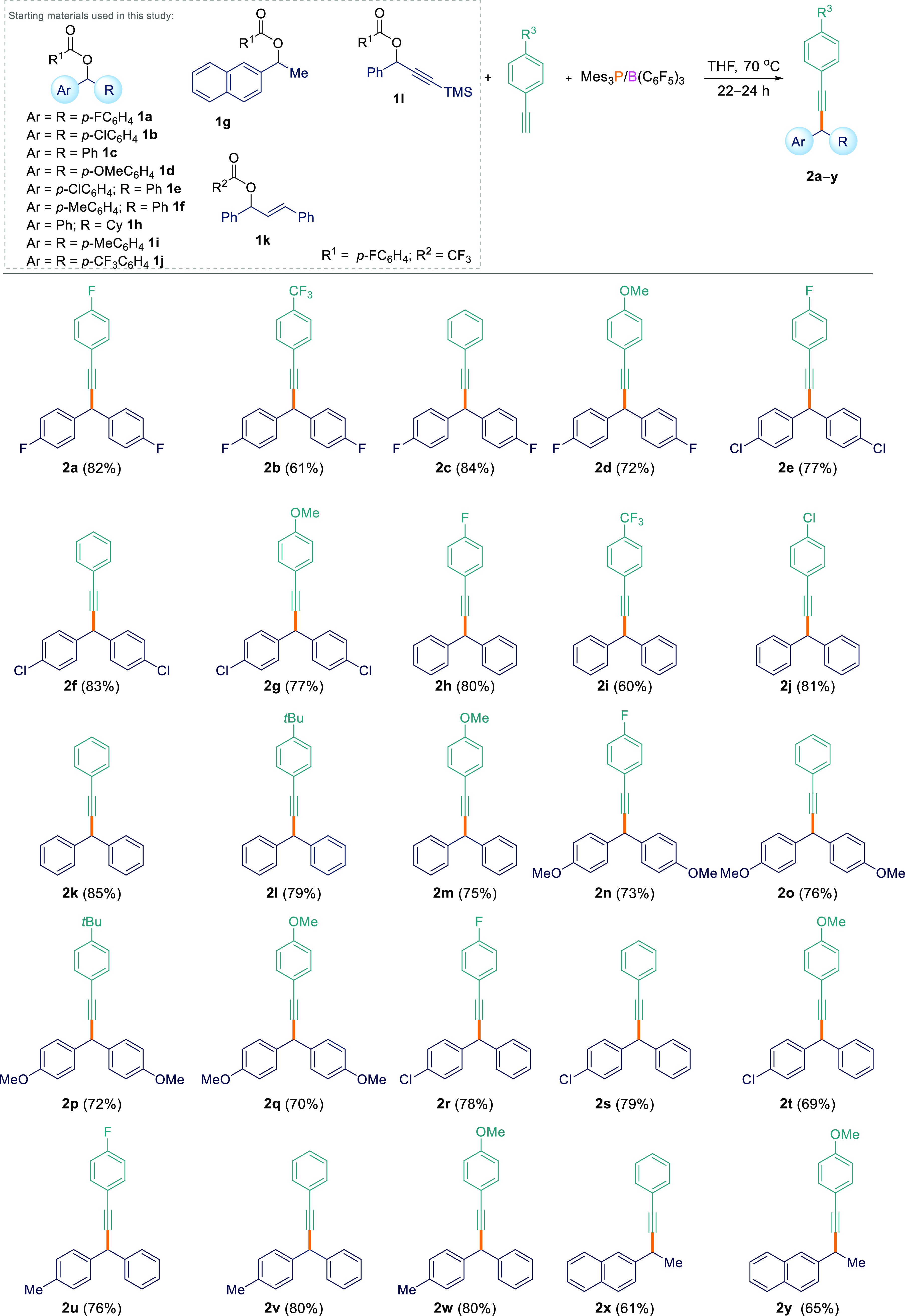
Csp3–Csp Cross-Coupling Reactions between Esters 1 and Acetylenes
All reactions were performed on a 0.1 mmol scale.
After achieving good success for the Csp3–Csp cross-coupling reaction at the benzylic position, we investigated a wider substrate scope (Scheme 3). To this end, allylic ester (E)-1,3-diphenylallyl-2,2,2-trifluoroacetate (1k) was used in the C–H functionalization. To our delight, excellent yields of products 2z–ah (72–89%) were obtained. While benzylic and allenylic esters worked well, the same was not true for cross-coupling at the propargylic position. When the aryl/alkynyl ester 1-phenyl-3-(trimethylsilyl)prop-2-yn-1-yl 4-fluorobenzoate (1l) was employed with terminal acetylenes, an inseparable complicated reaction mixture resulted.
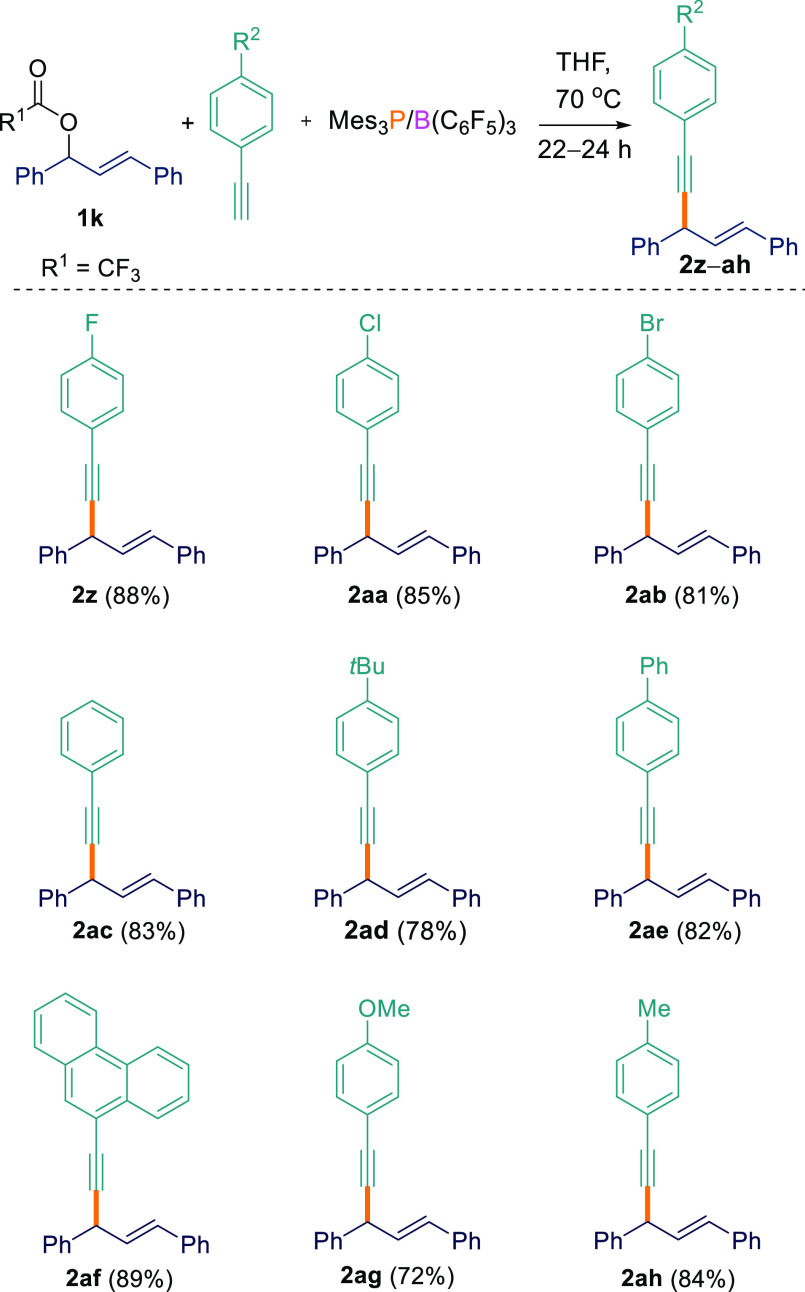
Csp3–Csp Cross-Coupling Reactions between Ester 1k and Acetylenes
All reactions were performed on a 0.1 mmol scale.
In our previous studies,6 we have shown that reactions of esters 1 with styrenes in the presence of the same FLP leads to Csp3–Csp2 coupled products. We therefore undertook an experiment to investigate the regioselectivity of the reaction by reacting the ester starting material with a 1:1 mixture of an acetylene and a styrene. For this reaction, three outcomes are theoretically possible: (i) formation of the Csp3–Csp coupled product, (ii) formation of the Csp3–Csp2 coupled product, or (iii) formation of a mixture of Csp3–Csp and Csp3–Csp2 coupled products. Using the optimized reaction conditions, an equimolar mixture of aryl ester 1a, 4-fluorophenylacetylene, and 4-fluorostyrene were reacted together with B(C6F5)3/Mes3P (Table 2).
| entry | R1 | R2 | solvent | yield Csp3–Csp (%) | yield Csp3–Csp2 (%) |
|---|---|---|---|---|---|
| 1 | F | H | THF | 78 (2a) | n.d. |
| 2 | F | H | TFT | 72 (2a) | n.d. |
| 3 | H | Ph | THF | n.d. | 63 (3a) |
| 4 | H | TMS | THF | 58 (2c) | 18 (3a) |
a 0.1 mmol scale, reported yields are isolated; n.d. = not detected.
The crude 1H NMR spectrum clearly showed a sharp singlet at δ = 5.09 ppm which confirmed the formation of the Csp3–Csp cross-coupled product, 2a, isolated as the major product in 78% yield (Table 2, entry 1). We were not able to detect any characteristic peaks (i.e., a doublet at δ = 4.98 ppm in the 1H NMR spectrum)6 for the Csp3–Csp2 coupled compound. To investigate the effect of solvent on the reaction, we also conducted the reaction in TFT, where again 2a was isolated as the major product albeit with a slightly reduced yield of 72% (Table 2, entry 2). The observation of exclusive Csp3–Csp coupling is presumably a consequence of the higher reactivity of the alkyne functionality over the alkene. A similar competition experiment, using a 1:1 mixture of styrene and the internal alkyne diphenylacetylene, gave only Csp3–Csp2 coupling producing 3a in 63% yield (Table 2, entry 3). Interestingly, TMS-protected alkynes behaved in the same manner as terminal alkynes, predominantly giving the Csp3–Csp coupled product with the loss of the TMS group. Using a 1:1 mixture of styrene and trimethyl(phenylethynyl)silane afforded the Csp3–Csp coupled product (2c, 58% isolated) as the major product with small amounts of the Csp3–Csp2 cross-coupled product (3a, 18% isolated) formed (Table 2, entry 4). This was also observed by in situ1H NMR spectroscopy of the crude reaction mixture, which displayed a 1:0.4 ratio of Csp3–Csp to Csp3–Csp2 cross-coupled products.
To demonstrate the scope for this selectivity, we synthesized a substrate containing both alkene and alkyne functionalities, namely, 1-ethynyl-4-vinylbenzene (4a).174a was subsequently reacted with different aryl esters (1a,c,e) in the presence of the B(C6F5)3/Mes3P FLP. In agreement with the observation above, we observed only the formation of the Csp3–Csp compounds as the major product (2ai–ak; 70–76%) using the optimized reaction conditions (Scheme 4). In all cases, the Csp3–Csp2 coupled product was either not detected or was observed in <5% yield in both THF and TFT solvents.
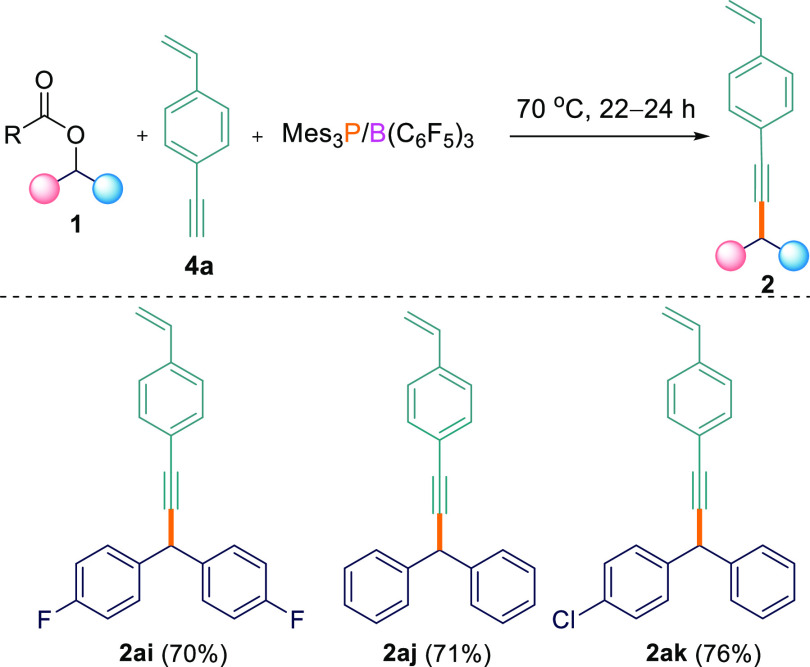
Cross-Coupling Reactions between Esters 1a,c,e and 1-Ethynyl-4-vinylbenzene 4a
0.1 mmol scale; reported yields are isolated.
As for the intermolecular competition reactions, we also synthesized internal alkynes in which the acetylenic proton in 4a was replaced by a phenyl or TMS group to explore how this affected the regioselectivity of the reaction (Scheme 5). Using the optimized reaction conditions, the reaction of aryl ester 1a with 1-(phenylethynyl)-4-vinylbenzene (4b) exclusively gave the Csp3–Csp2 cross-coupled product in 71% isolated yield from the reaction with the alkene, whereas when 1a was reacted with trimethyl{(4-vinylphenyl)ethynyl}silane (4c), reaction at the alkyne and removal of the TMS group in situ afforded the Csp3–Csp cross-coupled product (2ai) in 69% yield, with no significant reaction at the alkene site observed.
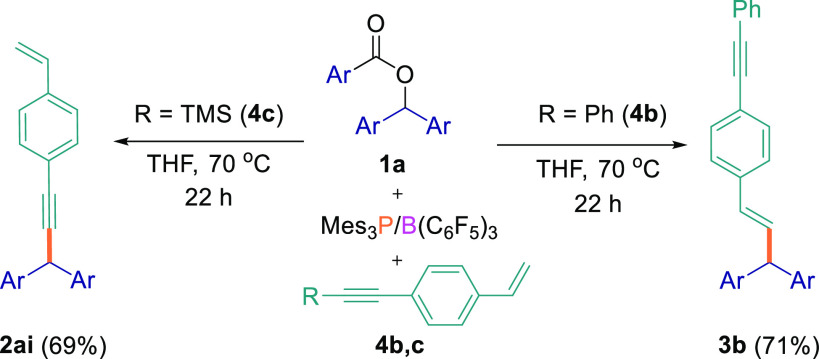
Cross-Coupling Reactions between Ester 1a and Substrates 4 bearing Internal Alkynes
All reactions were performed on a 0.1 mmol scale.
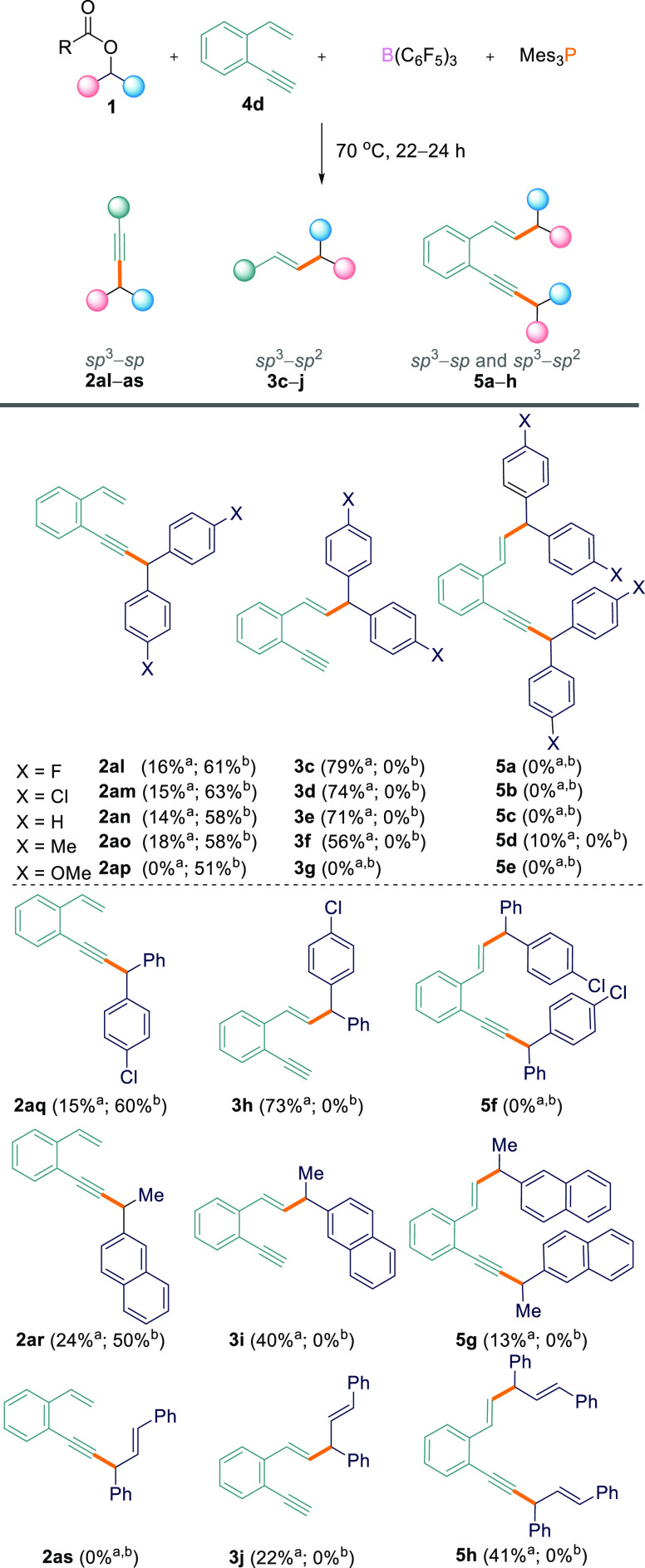
With these results in hand, we further explored the substrate scope using the 1-ethynyl-2-vinylbenzene (4d).184d was reacted with ester 1a and the B(C6F5)3/Mes3P FLP under the optimized reaction conditions (THF, 70 °C, 24 h). Contrary to the reactions above, the reactions were found to be highly site-selective for the Csp3–Csp2 coupled product, 3, from reaction at the alkene functional group. Examining the crude 1H NMR spectrum revealed a 0.2:1:0 ratio of the products (Csp3–Csp coupled, 2)/(Csp3–Csp2 coupled, 3)/(Csp3–Csp and Csp3–Csp2 coupled, 5) (Table 3, entry 1) with isolated yields of 79% for the Csp3–Csp2 coupled product, 3c, and 16% for the Csp3–Csp coupled product, 2al (Scheme 6). Interestingly, when changing the solvent to TFT, the selectivity was completely reversed, exclusively giving Csp3–Csp product 2al from reaction at the alkyne site.
Products of Solvent-Dependent Site-Selective Cross-Coupling Reactions
Reactions were carried out on a 0.1 mmol scale, and yields were isolated. aYield in THF. bYield in TFT.
| entry | ester | ratio of 2:3:5 in THF | entry | ester | ratio of 2:3:5 in TFT |
|---|---|---|---|---|---|
| 1 | 1a | 0.2:1:0 | 9 | 1a | 1:0:0 |
| 2 | 1b | 0.2:1:0 | 10 | 1b | 1:0:0 |
| 3 | 1c | 0.2:1:0 | 11 | 1c | 1:0:0 |
| 4 | 1i | 0.4:1:0.1 | 12 | 1i | 1:0:0 |
| 5 | 1d | 0:0:0 | 13 | 1d | 1:0:0 |
| 6 | 1e | 0.2:1:0 | 14 | 1e | 1:0:0 |
| 7 | 1g | 0.5:1:0.1 | 15 | 1g | 1:0:0 |
| 8 | 1j | 0:1:1.5 | 16 | 1j | 0:0:0 |
a Ratios were determined from the crude 1H NMR spectra.
This was observed by crude 1H NMR, indicating a 1:0:0 ratio of 2/3/5 (Table 3, entry 9) in which 2al could be isolated in 61% yield (Scheme 6). Remarkably, by simply changing the solvent we can switch the site selectivity of the reaction.
We next investigated this solvent-dependent site selectivity for a range of other esters and found the same general trend. In the following discussion, all reaction product ratios were determined via crude 1H NMR studies and are listed in Table 3, with the corresponding isolated yields for the products shown in Scheme 6. Initially, we explored the reactions in THF solvent. When electron-withdrawing 1b (p-Cl) or electron-neutral symmetrical diaryl esters 1c (p-H) and 1i (p-Me) were used, there was a clear preference for reaction at the alkene site leading to Csp3–Csp2 coupled products 3d, 3e, and 3f in ratios of 0.2:1:0, 0.2:1:0, and 0.4:1:0.1 for 2/3/5, respectively (Table 3, entries 2–4). In all cases, the major and minor regioisomers could be separated. The Csp3–Csp2 cross-coupled products were isolated in 74% (3d), 71% (3e), and 56% (3f) yields, and the Csp3–Csp cross-coupled products were isolated in 15% (2am), 14% (2an), and 18% (2ao) yields. In the case of 1i as the starting material, double cross-coupled product 5d was observed in small amounts and could be separated and isolated in 10% yield (Scheme 6). Electron-rich p-OMe ester 1d, on the other hand, showed no reactivity at all in THF (Table 3, entry 5). Asymmetrical diaryl ester 1e gave a ratio of 0.2:1:0 for 2/3/5 (Table 3, entry 6), with 2aq and 3h being isolated in 15 and 73% yields, respectively. Alkyl/aryl ester 1g also gave the Csp3–Csp2 cross-coupled product as the major isomer, albeit less selectively, showing a 0.5:1:0.1 ratio of the three products 2ar/3i/5g in isolated yields of 24% (2ar), 40% (3i), and 13% (5g) (Table 3, entry 7 and Scheme 6). 1,3-Diphenylallyl-2,2,2-trifluoroacetate (1k), on the other hand, showed a preference for the double cross-coupled product, giving a 0:1:1.5 ratio of 2/3/5 with isolated yields of 22% (3j) and 41% (5h) (Table 3, entry 8 and Scheme 6).
Subsequently, we repeated the above series of reactions in TFT, and remarkably, the regioselectivity was altered and the selectivity was improved. No Csp3–Csp2 coupled product (3) or double Csp3–Csp/Csp3–Csp2 coupled product (5) was observed with any combination of substrates. Rather, a ratio of 1:0:0 of products 2/3/5 was observed in all cases for esters 1a–e, 1g, and 1i (Table 3, entries 9–15). This included p-OMe ester 1d which did not show any product formation in THF. Csp3–Csp cross-coupled products 2al–ar could be isolated in 50–63% yields (Scheme 6). The only exception was 1,3-diphenylallyl 2,2,2-trifluoroacetate (1k), which gave a complex mixture of inseparable products when reacted with 1-ethynyl-2-vinylbenzene (4d) in TFT, none of which could be identified as 2, 3, or 5 (Table 2, entry 16).
Proposed Reaction Mechanism
The Csp3–Csp cross-coupling reaction could be explained by either a single- or a two-electron pathway.
First, a diamagnetic pathway could operate (Scheme 7A) in which the Lewis acid component of the FLP activates the ester carbonyl atom, leading to the formation of diaryl methylene cation I3 and the borate anion [R1CO2B(C6F5)3]−. This was observed in our previous studies when B(C6F5)3 was added to the diaryl ester in the presence of a nucleophile to trap the resultant carbocation.19 The diaryl methylene cation and the Lewis basic component of the FLP can then undergo a 1,2-FLP addition to the alkyne similar to other FLP 1,2-additions.20 Finally, the elimination of [Mes3PH]+ leads to the C–C-bonded product and salt [R1CO2B(C6F5)3][HPMes3] (R1 = p-FC6H4 or CF3). This can be observed in the reaction by multinuclear NMR spectroscopy showing a clear 1JPH coupling of 479.5 Hz.
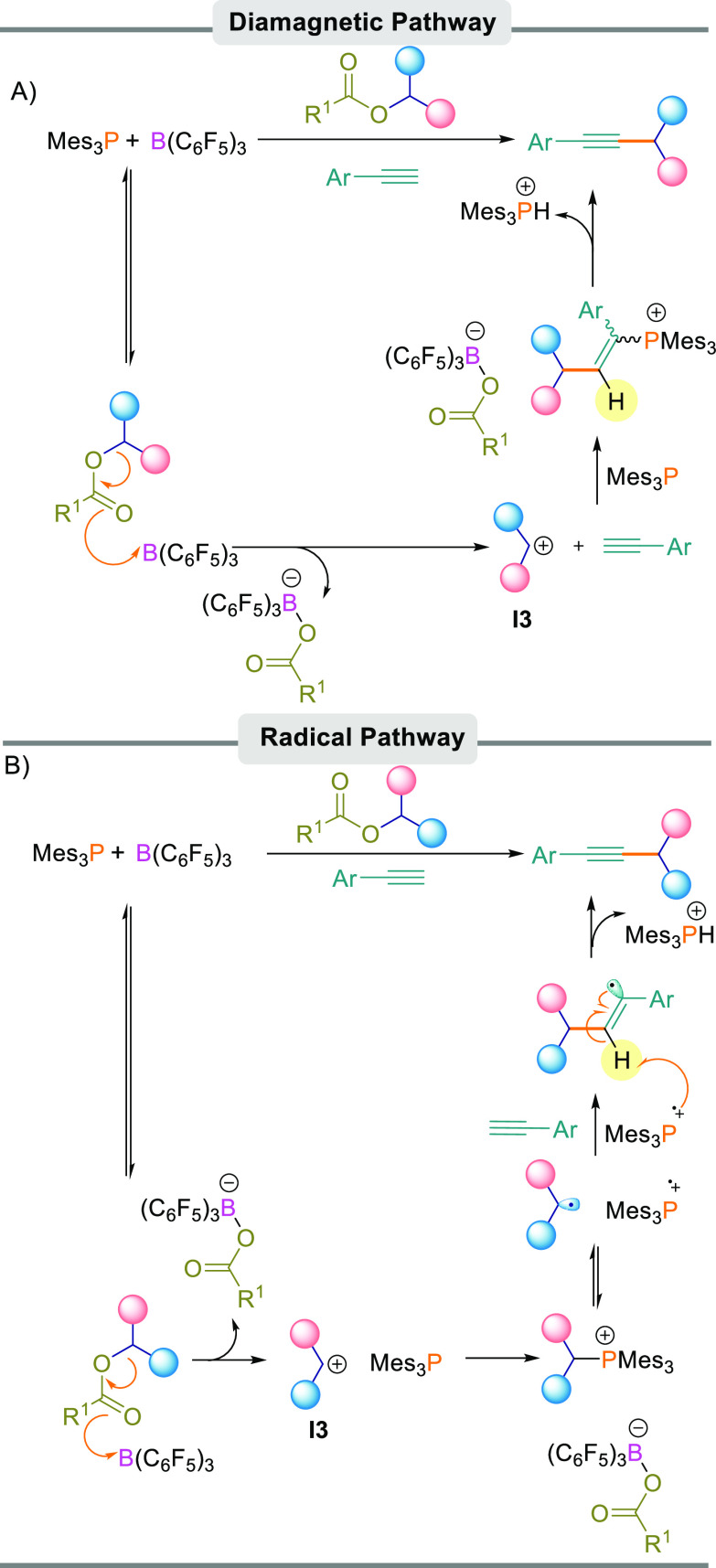
Possible Reaction Mechanisms
(A) Diamagnetic pathway. (B) Radical pathway.
Alternatively, a radical pathway could operate (Scheme 7B), which may explain the necessity for using Mes3P as a Lewis base rather than other phosphine or nitrogen bases. Previous studies have postulated that the B(C6F5)3/Mes3P FLP can undergo a single electron transfer (SET) process generating radical ion pair [B(C6F5)3]•–/[Mes3P]•+.21 Slootweg et al. later postulated that this process is promoted upon irradiation with visible light (390–500 nm).5b In this pathway (Scheme 7B), we propose that the first step of the reaction is identical to the diamagnetic pathway whereby B(C6F5)3 activates the diaryl ester to generate diaryl methylene cation I3 and borate anion [R1CO2B(C6F5)3]−. The Lewis base then reacts with cation I3, forming a Lewis acid–base adduct. We propose that this adduct is in equilibrium with diaryl methylene radical [Ar2CH]• and phosphonium radical cation [Mes3P]•+, which are formed from the homolytic cleavage of the C–P bond. From here, diaryl methylene radical [Ar2CH]• adds to arylacetylene, leading to a vinylic radical species which, upon abstraction of a hydrogen atom by [Mes3P]•+, generates the desired C–C bonded product.
To understand which pathway is operating, we undertook extensive electron paramagnetic resonance (EPR), kinetic, and density functional theory (DFT) studies to understand the reaction mechanism for the Csp3–Csp coupling reaction.
EPR Studies
The knowledge that the B(C6F5)3/Mes3P FLP can generate radical species prompted us to undertake an EPR study to determine if radical species could be observed in these reactions. As reported previously, no EPR signal could be detected from the B(C6F5)3/Mes3P FLP in the absence of any substrates.21 Upon addition of an equimolar ratio of ester 1d to the FLP in a TFT solution, several EPR signals arising from multiple paramagnetic species were detected at room temperature (Figure 1A). Upon comparison with previous reports, the two intense resonance lines in a 1:1 ratio (marked with asterisks) centered on giso = 2.012 (B ≈ 335 mT) and separated by a phosphorus hyperfine splitting of aiso(31P) = 670 MHz (23.8 mT) are attributed to the formation of the [Mes3P]•+ cation.22 A second paramagnetic species was also detected in this sample, which is characterized by a 1:2:1 triplet centered on giso = 2.010 and separated by a hyperfine splitting of 470 MHz (16.7 mT). This EPR profile must originate from two identical I = 1/2 nuclei, in this case associated with two equivalent 31P nuclei. The signal is therefore tentatively assigned to the formation of a [(P(Mes)n=2,3)2]•+ dimer formed from the association of excess Mes3P with radical cation [Mes3P]•+ as observed previously.23 This assignment is supported by our observation that the relative ratios of the EPR signals of [Mes3P]•+/[(P(Mes)n=2,3)2]•+ are inter-related (i.e., when a large signal intensity of the monomer is observed, only a trace of the corresponding dimer is detected). The varying ratio of these EPR signals under different reaction conditions demonstrates the conversion between monomer and dimer via the reaction of the monomer with a second molecule of phosphine to yield the dimer radical cation, as previously observed for a series of phosphines,24 and possibly also the varying stabilities of the two radical species. The EPR spectrum of [(PMes2)2]•+ has previously been reported,25 characterized by giso = 2.006 and aiso(31P) = 474 MHz (17 mT), and it is noted that literature examples of phosphine dimer cation radicals of divalent (R2P)2•+ and trivalent (R3P)2•+ systems yield very similar EPR spectra,26 dominated by the 1:2:1 phosphorus hyperfine splitting.
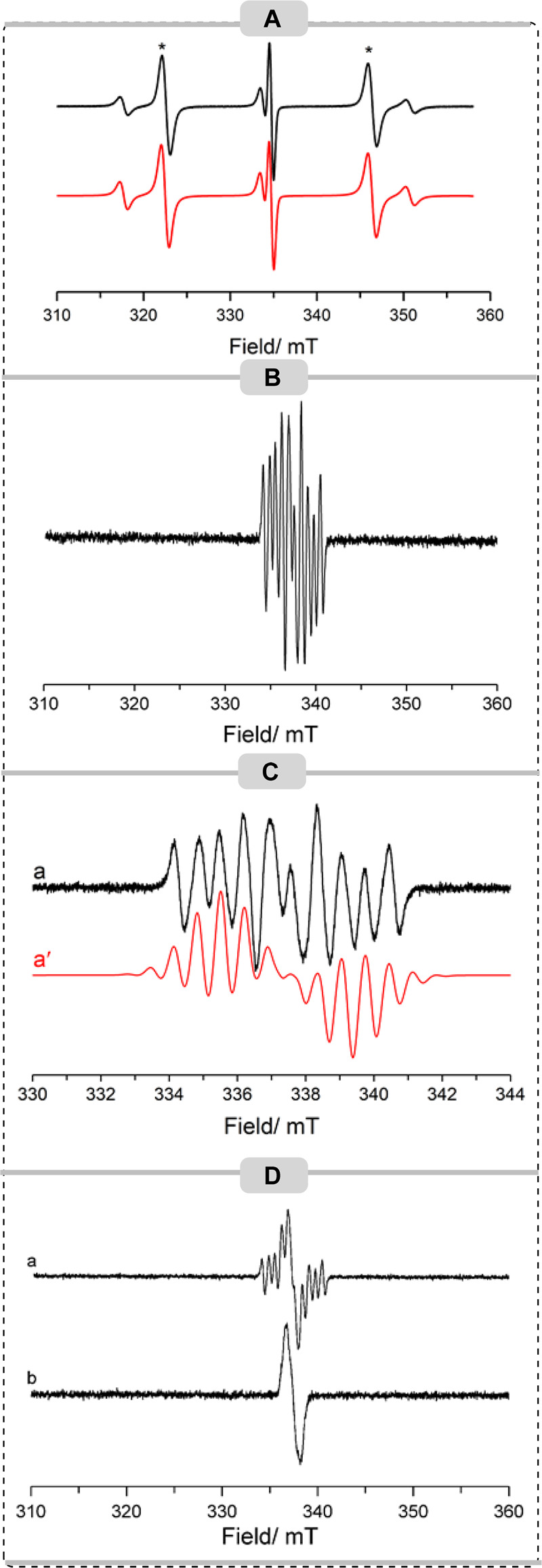
CW X-band EPR spectra (T = 298 K) of (A) FLP + ester 1d, (B) FLP + phenylacetylene, (C) FLP + phenylacetylene (black, experimental; red, simulated), and (D) FLP + phenylacetylene + ester recorded at (a) t = 0 min and (b) after storage at 77 K for 30 min. TFT was used as the solvent for all EPR measurements.
The corresponding anisotropic EPR spectra of the [Mes3P]•+ monomer and [P(Mes)n=2,3]2•+ dimer species in frozen solutions are shown in the SI (Figure S176), from which the principal values of the g and A tensors were determined. The spin Hamiltonian parameters for all of the paramagnetic species detected in this work are listed in the SI (Table S1). Importantly, contrary to our previous reports,6 no evidence for the formation of the carbon-based bismethoxy-diphenylmethylene radical formed upon C–O bond cleavage was observed in this case, perhaps due to the instability of the radical species.
We then probed the room-temperature EPR spectrum of the FLP in the presence of phenylacetylene (Figure 1B,C). Under these experimental conditions, we postulated that another possible mechanism could be the abstraction of the terminal hydrogen atom of the acetylene by the [Mes3P]•+ radical cation to form the diamagnetic [Mes3PH]+ cation and the corresponding phenylacetylene radical. As can be seen from the wide field scanning range in Figure 1B, no evidence of monomer [Mes3P]•+ or dimer [P(Mes)n=2,3]2•+ was observed in this solution, suggesting the formation of the diamagnetic [Mes3PH]+ cation. However, no EPR evidence for the generation of the phenylacetylene radical was obtained. It is noted in previous literature studies that the terminal phenylacetylene radical is inherently unstable and is typically observed only via EPR spectroscopy under controlled conditions, such as neat liquids sealed under vacuum, or via matrix isolation methods.27 Notably, the remaining boron component of the FLP is not involved in the above hydrogen atom abstraction from the alkyne and may be expected to remain in solution. The intense multiplet signal observed, reproduced in Figure 1C across a narrow field range, is therefore assigned to the boron radical anion, [B(C6F5)3]•–. A satisfactory simulation of the experimental data was achieved using giso = 2.0114 and incorporating a single boron nucleus, aiso(10,11B) = 27 MHz, two sets of six equivalent fluorine nuclei from the ortho and meta positions, with aiso(19F)ortho = 18.28 MHz and aiso(19F)meta = 3 MHz, and three para fluorine nuclei with aiso(19F)para = 20.2 MHz, which agrees well with previous literature reports of this species.28 The corresponding anisotropic spectrum for this sample was unfortunately not resolved due to a poor-quality glass of the frozen solvent, thereby preventing the determination of the complete anisotropic spin Hamiltonian parameters for this radical anion.
Having determined the reactivity of the FLP to the individual substrates, the EPR spectrum of a full reaction mixture containing equimolar amounts of Mes3P, B(C6F5)3, ester, and phenylacetylene was recorded (Figure 1Da). As can easily be seen, evidence of the [B(C6F5)3]•– radical anion is clearly observed, but there are no signals attributed to monomer or dimer phosphorus radicals present. However, it is noticed that there is additional intensity superimposed in the center of this signal (giso ≈ 2.001) which must originate from a second paramagnetic species not previously observed. Upon storage of this sample at 77 K for 30 min, the signal intensity originating from the [B(C6F5)3]•– radical anion was lost completely, leaving only a narrow resonance (Figure 1Db). Unfortunately, the short lifetime of this radical in solution prevented full resolution of the hyperfine coupling, but the narrow spectral width arising from only small hyperfine couplings is an indication of a carbon-based radical rather than a boron or phosphorus species (upon consideration of the theoretical isotropic hyperfine a0 values a0(10B) = 30.43 mT, a0(11B) = 90.88 mT, and a0(31P) = 474.79 mT). The experimental spectrum is reproduced again in the SI (Figure S177) alongside simulations of the styrene and phenylacetylene radicals, using previously reported literature values.29 Gratifyingly, there is reasonable agreement between the experimental and simulated data, thereby this signal is tentatively attributed to a carbon-based radical, perhaps indicating that the reaction could be occurring through a radical mechanism.
DFT and Kinetic Studies
To examine the contrasting reaction pathways and to explain the experimental and EPR observations, we undertook a thorough DFT investigation of all potential reaction pathways. Calculations were performed at the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d) level of theory in THF and toluene solvent to examine the origin of the products. As previously reported by Slootweg et al., we found that the formation of the frustrated radical ion pair from the FLP is energetically unfavorable by 35.7 kcal/mol. This corroborates the observation that we and others6 fail to see any EPR signal in solutions of B(C6F5)3 and Mes3P. Very recently, Slootweg et al.5a showed that the coordination of B(C6F5)3 to the diaryl ester increases the electron affinity of the substrate, and the energy required for SET from Mes3P to the methylene carbon atom is 40.0 kcal/mol.5a This is still quite large, and our calculations have revealed that, regardless of whether a diamagnetic or paramagnetic reaction pathway is ultimately operative, the first step in the reaction is the same: B(C6F5)3 activation of the ester to generate diaryl methylene cation I3 (Scheme 8) and the borate anion [R1CO2B(C6F5)3]− with an activation energy of 10.7 kcal/mol. (See SIFigure S178 for the free-energy profile.) The energies of I3 can be noticeably varied by changing the substitution at the para position of the aryl esters. Electron-withdrawing (p-CF3, 1j), electron-withdrawing/π-releasing (p-F, 1a), and electron-releasing (p-OMe, 1d) showed different energies for I3 (SIFigure S179–180). As expected, I31d (−9.4 kcal/mol) is energetically more favorable than I31j (13.5 kcal/mol) due to the electron-releasing substituents (p-OMe). Strongly electron-withdrawing substituents (p-CF3) conversely make I3 formation thermodynamically less favorable. I3 is then the branching point for the single- and two-electron pathways (Figure 2). The combination of diaryl methylene cation I3 with Mes3P can lead to three possible species in solution (Scheme 8): (i) the frustrated Lewis pair (uncoordinated I3 + Mes3P), (ii) the Lewis acid–base adduct (I4), and (iii) the frustrated radical pair (FRP, I5). The energy difference and reaction barriers between these species are very low; therefore, it is likely that all three scenarios exist in equilibrium under the reaction conditions. This supports the EPR data which shows the formation of [Mes3P]•+ and the [(P(Mesn=2,3)2]•+ dimer in the reaction of the ester with the Mes3P/B(C6F5)3 FLP.
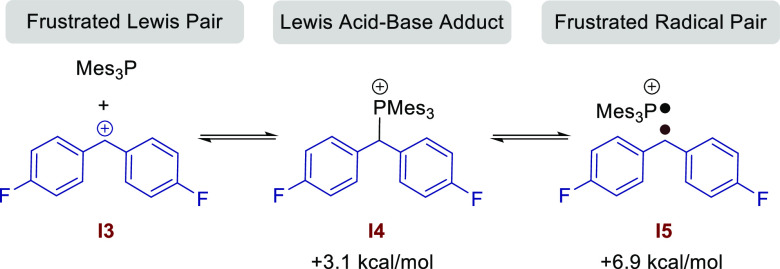
Mes3P/Diaryl Methylene Cation Equilibria in Solution
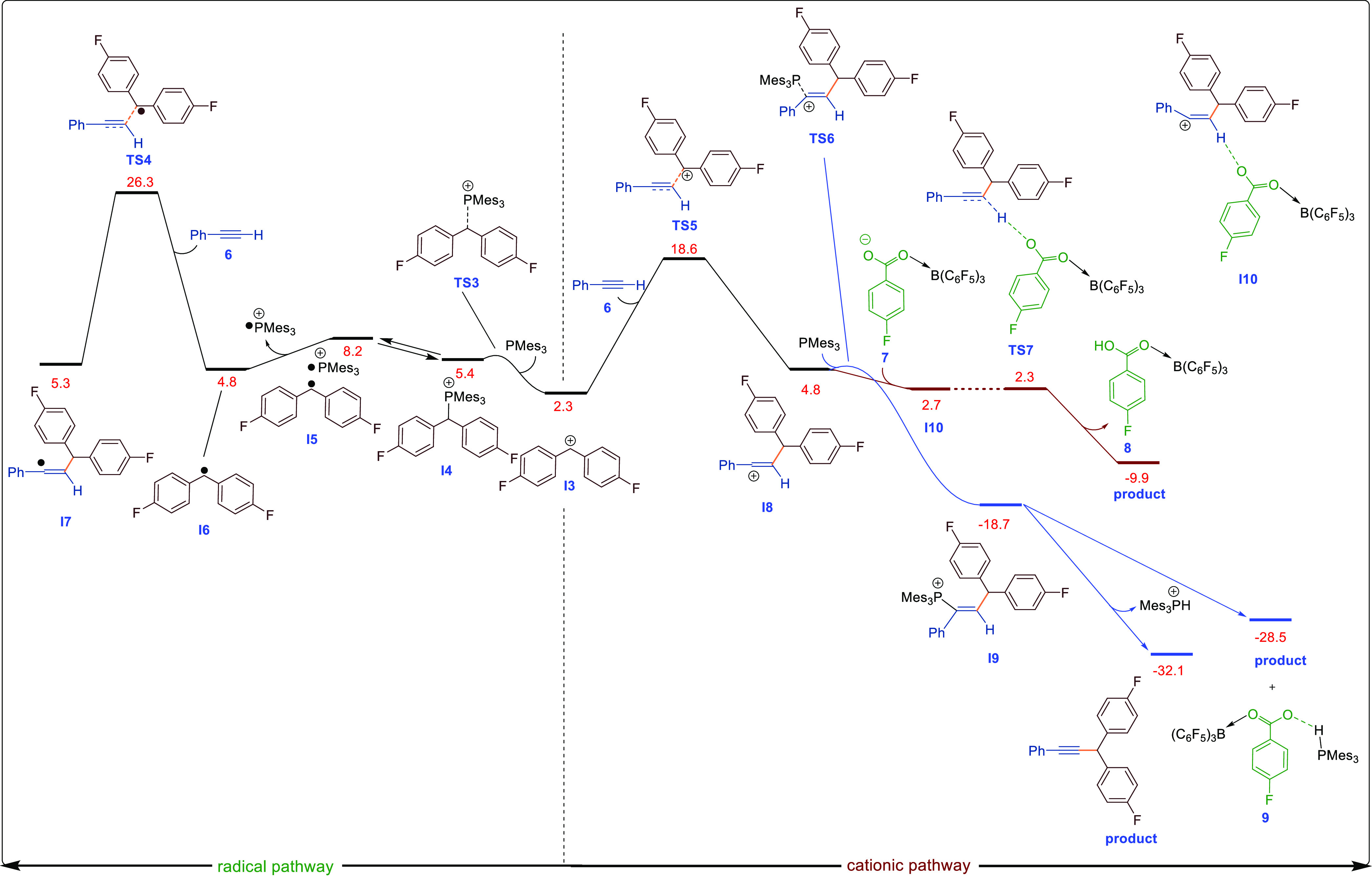
DFT-computed reaction pathways for the reaction of I3 with phenylacetylene calculated by SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d) in THF. The relative free energies are given in kcal/mol. For this energy profile, structure 1a is set as the reference point as indicated in Figure S178.
Although the carbon-based radical could not be observed in this case, we have observed a weak carbon-based radical EPR signal when reacting other esters with the Mes3P/B(C6F5)3 FLP in the absence of irradiation or heat.6 We then investigated the addition of the cation (I3) or radical (I6) to the alkyne. Although both pathways are feasible under the reaction conditions, the cationic pathway was lower in activation energy than the radical pathway by about 26.3–18.6 = 7.7 kcal/mol. In the case of the diamagnetic pathway, the addition of the I3 cation to the alkyne generates I8 via TS5. The resulting cation in I8 is highly reactive and is rapidly trapped by the Lewis base Mes3P generating I9. Finally, anti-elimination of [Mes3PH]+ generates the cross-coupled compound and phosphonium borate salt as the final products.
The Lewis base employed is very important for the reaction to occur as seen in the screening studies. First, the ability to form an FLP (or weak adduct) with both the Lewis acid borane and the (di)aryl methylene cation (I3) is critical, as other strong, less hindered Lewis acids such as BF3 do not work in the reaction. Likewise, smaller phosphines or amines tend to coordinate more strongly to the carbocation (I3). In addition to being able to form an FLP, the base also functions to trap reactive I8. Indeed, one could possibly conceive that the reaction could proceed with the Lewis acid only, whereby R1CO2– accepts the proton in the last step (I8 → I10 → product, Figure 2). However, DFT calculations showed that this pathway was less favorable thermodynamically, and experimentally this reaction showed only a 22% yield with many side products formed.
We were curious to know whether both the paramagnetic and diamagnetic pathways are operative in parallel or if a diamagnetic mechanism is purely responsible for the product formation for all alkynes and the radicals observed from EPR studies were simply off-pathway intermediates. To establish this, we undertook further DFT calculations to investigate the effect of electron-withdrawing, electron-donating, and electron-neutral substituents on the ester (1) and the alkyne on the activation barrier for the reaction.
The energy barriers TS4radical pathway and TS5cationic pathway were calculated (Table 4, see SIFigure S181). Initially we varied the electronic properties of the alkyne using p-XC6H4C≡CH (X = NO2, CF3, H, OMe, NMe2) with ester 1a. As evidence from the DFT calculations, changing the substitution at the para position of the arylacetylene did not make significant difference for TS4radical pathway in their respective energy barrier (23.9–27.0 kcal/mol) (Table 4, entries 1–5). However, the energy barrier for TS5cationic pathway changed dramatically (8.2–23.2 kcal/mol). When electron-withdrawing groups (p-NO2 and p-CF3) on the arylacetylene were employed, the differences between TS4radical pathway and TS5cationic pathway are 0.7 and 6.1 kcal/mol (Table 4, entries 1 and 2). Electronically neutral phenylacetylene exhibits a TS4radical pathway → TS5cationic pathway difference of 7.7 kcal/mol (Table 3, entry 3). Electron-donating groups such as methoxy (TS4radical pathway – TS5cationic pathway = 12.1 kcal/mol) and N,N-dimethylamine (TS4radical pathway – TS5cationic pathway = 18.8 kcal/mol), on the other hand, showed a significant energy difference (Table 4, entries 4 and 5). These observations can be seen in Figure 3, which shows that for TS4 there is a negligible change in the energy barrier when changing the electronic properties of the acetylenic substrate. This is in agreement with little charge formation on the reaction center in the radical mechanism. Conversely, for TS5, there is a strong positive correlation between the TS5 energy barrier and the substituent σp constant. This is expected for the cationic pathway because a developing positive charge adjacent to the substituted phenyl ring will be stabilized by electron-donating groups (e.g., p-NMe2 or p-OMe) on the acetylene. As can be seen in Figure 3, TS5 is lower than TS4 for all substituents explored, although the difference becomes small for strongly electron-withdrawing groups.
| entry | ester (Ar) | X | TS4radical pathway | TS5cationic pathway |
|---|---|---|---|---|
| 1 | 1a (p-FC6H4) | NO2 | 23.9 | 23.2 |
| 2 | 1a (p-FC6H4) | CF3 | 26.2 | 20.1 |
| 3 | 1a (p-FC6H4) | H | 26.3 | 18.6 |
| 4 | 1a (p-FC6H4) | OMe | 26.0 | 13.9 |
| 5 | 1a (p-FC6H4) | NMe2 | 27.0 | 8.2 |
| 6 | 1d (p-OMeC6H4) | CF3 | 23.7 | 13.0 |
| 7 | 1d (p-OMeC6H4) | OMe | 27.8 | 8.1 |
| 8 | 1j (p-CF3C6H4) | CF3 | 24.1 | 25.5 |
| 9 | 1j (p-CF3C6H4) | OMe | 22.2 | 17.5 |
a The relative free energies are given in kcal/mol.
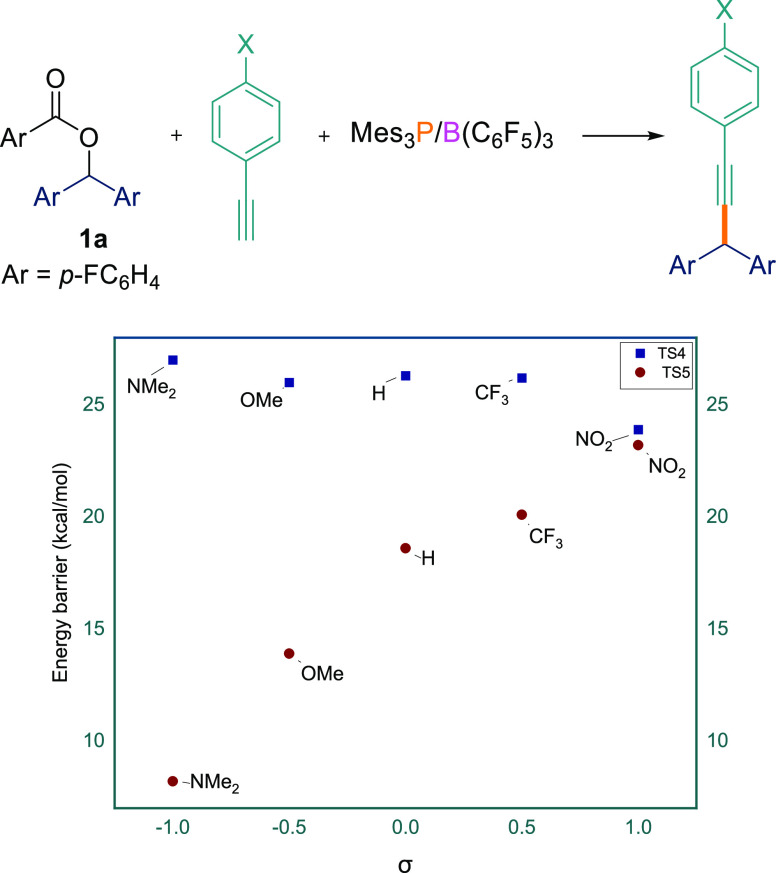
We subsequently computed the energy barrier for the two transition states by varying the electronic properties of the aryl ester using electron-donating (p-OMe, 1d) and electron-withdrawing (p-CF3, 1j) esters with electron-deficient (1-ethynyl-4-(trifluoromethyl)benzene) and electron-rich (1-ethynyl-4-methoxybenzene) acetylenes (Table 4, entries 6–9). For both esters, a smaller change in the TS4radical pathway energy barrier was observed (range 22.2–27.8 kcal/mol) compared to the TS5cationic pathway energy barrier (range 8.1–25.5 kcal/mol) when changing the substituent on the phenylacetylene (Table 4, entries 6–9). As with ester 1a, both esters disclosed a larger energy difference between the two pathways when reacted with acetylenic compounds bearing an electron-donating group (p-OMe). Likewise, a much smaller energy difference was noted for both esters when reacted with acetylenic compounds bearing an electron-withdrawing group (p-CF3). Interestingly, DFT studies showed that for the case of the reaction of ester 1j with 1-ethynyl-4-(trifluoromethyl), the radical pathway is slightly energetically more favorable compared with the cationic pathway (Table 4, entry 8). These results suggest that for electron-withdrawing arylacetylenes both paramagnetic and diamagnetic mechanisms are potentially possible, whereas for electron-rich arylacetylenes a purely diamagnetic pathway is operative.
The key difference in the two mechanisms involves the reaction of either a cationic intermediate or a radical intermediate with the arylacetylene, generating a new cationic or radical species. Whether this new intermediate is a cationic or a neutral radical species can be probed using a Hammett plot (cf. computational studies) based on substituted arylacetylenes. To gain this insight into the reaction pathway and substituent effects also from experimental evidence, we examined competition reactions among the FLP, aryl ester 1a, and arylacetylenes p-XC6H4C≡CH bearing electron-withdrawing, electron-neutral, and electron-releasing groups. The Hammett plot requires relative rate constants for the reaction of different substituted alkynes that were obtained using a series of competition experiments. Initial competition experiments in the presence of 1.5 equiv of five arylacetylenes p-XC6H4C≡CH (X = CF3, F, Cl, H, and OMe) were unsuccessful. The excess arylacetylene present in the reaction mixture destroyed the efficacy of the FLP system, producing a complicated reaction mixture which was not suitable for in situ NMR analysis. We therefore carried out three binary competition experiments with two alkynes being present at 1.5 equiv each (Table 5).
| entry | X | X′ | product ratio | kx/kx′ |
|---|---|---|---|---|
| 1 | OMe | H | (2d:2c) 14.3:1 | 21.9 |
| 2 | CF3 | H | (2b:2c) 0.12:1 | 0.082 |
| 3 | OMe | CF3 | (2d:2b) 1: < 0.05 |
Using the optimized reaction conditions, three parallel reactions were carried out in which equimolar mixtures of (a) 1-ethynyl-4-methoxybenzene and phenylacetylene, (b) 1-ethynyl-4-(trifluoromethyl)benzene and phenylacetylene, and (c) 1-ethynyl-4-methoxybenzene and 1-ethynyl-4-(trifluoromethyl)benzene were reacted with 1 equiv of ester 1a. The ratios of Csp3–Csp cross-coupled products (2d/2c, 2b/2c, and 2d/2b) were determined from the crude reaction mixture using 19F NMR spectroscopy (Table 5, see SI, Figure S175). For entries 1 and 2 in Table 5, the relative integrals for the products were used to calculate the remaining equivalents for the alkynes after reaction. Using the approach developed by Ingold and Shaw30 and proposed for one-pot Hammett plots by Harper and co-workers,31 we obtained relative rate constants kx/kx′ for the reaction of differently substituted arylacetylenes with reaction intermediate I3 or its equilibrium species. Entry 3 confirms that in the competition between 1a and 1-ethynyl-4-(trifluoromethyl)benzene/1-ethynyl-4-methoxybenzene, product 2b is undetectable, in agreement with the >200-fold difference in rate constants deduced from entries 1 and 2. The relative rate constants in Table 5 are normalized with respect to the unsubstituted alkyne and can therefore be used directly to construct a Hammett plot (using Hammett substituent constants from ref (32)). The Hammett plot (Figure 4) shows a clearly negative slope of −6.6 ± 1.7, suggesting the formation of a positive charge on the new intermediate, which is indicative of the cationic reaction mechanism being operative. The Hammett plot is also in agreement with the Hammett-style plot constructed using the computational data (Figure 3) for the cationic mechanism. The slope of the Hammett-style plot for TS5 (Figure 3) is 8.9 ± 1.2 kcal mol–1. At 70 °C, this corresponds to a Hammett ρ value of 5.7 ± 0.8. The computational data is thus in excellent agreement with the experimental findings.
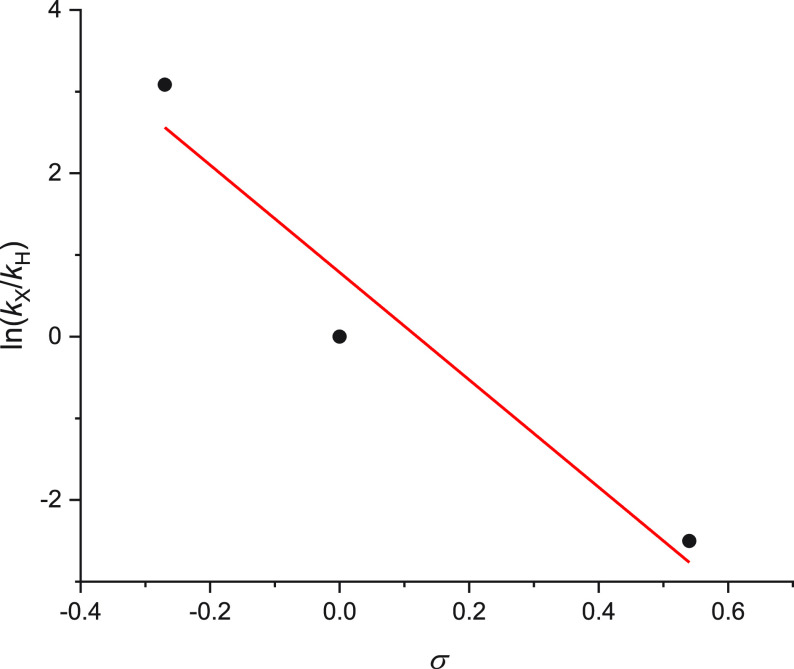
Hammett plot for the reaction of I3 with arylacetylenes.
Finally, we turned our attention to explaining the regioselectivity with compound 4a. The DFT-computed (SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d)) reaction pathways for the reaction of I3 with 1-ethynyl-4-vinylbenzene (4a) and Mes3P in THF reveal that the cationic pathway is energetically more favorable (Figure 5). After the generation of I3, reaction at either the alkyne or alkene site affords the corresponding Csp3–Csp or Csp3–Csp2 cross-coupled product. Although the transition-state energies for the I3-alkyne adduct (14.4 kcal/mol) and I3-alkene adduct (14.3 kcal/mol) are very similar, the Csp3–Csp cross-coupled products are thermodynamically more stable (−1.4 kcal/mol) than the Csp2–Csp2 cross-coupled product (5.7 kcal/mol), explaining why only the Csp3–Csp cross-coupled product is observed for 1-ethynyl-4-vinylbenzene (4a) (Figure 5). To confirm this, we also executed single-point benchmark calculations for this transition state with a different method and solvent system (SI, Table S2), which showed results similar to those indicated in Figure 5.
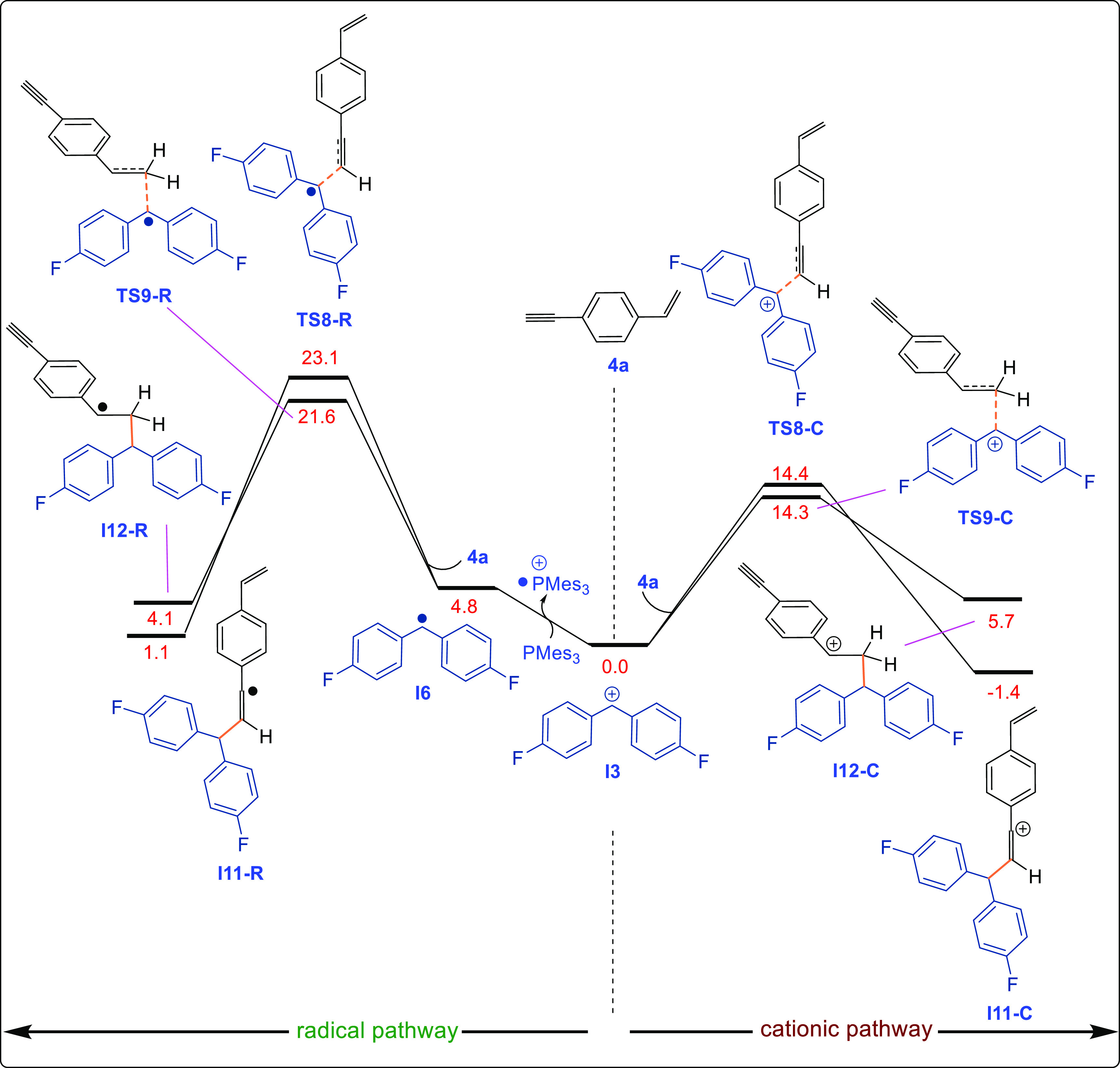
DFT-computed reaction pathways for the reaction of I3 with 1-ethynyl-4-vinylbenzene (4a) and Mes3P calculated using the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d) level of theory in THF.
The alternating site selectivity when using 1-ethynyl-2-vinylbenzene (4d) in differing solvents can also be highlighted in DFT studies. For the calculations, the mechanism was studied by utilizing two different solvents (toluene and THF). Experimentally, both toluene and TFT solvents lead to preferential Csp3–Csp coupling. DFT calculations for the reaction of I3 with 4d showed that the transition-state energy for the addition of the diaryl methylene cation to the alkene or alkyne varies depending upon the solvent (Figure 6). In toluene solvent, the energy barrier for the addition of I3 to the more nucleophilic alkyne was 3.7 kcal/mol lower in energy than the transition state for addition to the alkene (11.7 versus 15.4 kcal/mol). The converse was true for reactions in THF, whereby the addition of I3 to the alkene was 0.5 kcal/mol lower in energy than the energy barrier for addition to the alkyne (14.7 versus 15.2 kcal/mol). We attribute this to the higher dipole moment of the calculated transition-state structure of TS3a, which can be stabilized better by the THF molecule. These results also explain why, in THF, the reactions were less selective, leading to a mixture of Csp3–Csp and Csp3–Csp2 products due to the small energy difference between the two pathways. The reactions in a solvent such as toluene (or TFT) are more selective toward Csp3–Csp coupling due to the larger energy difference between the two pathways.
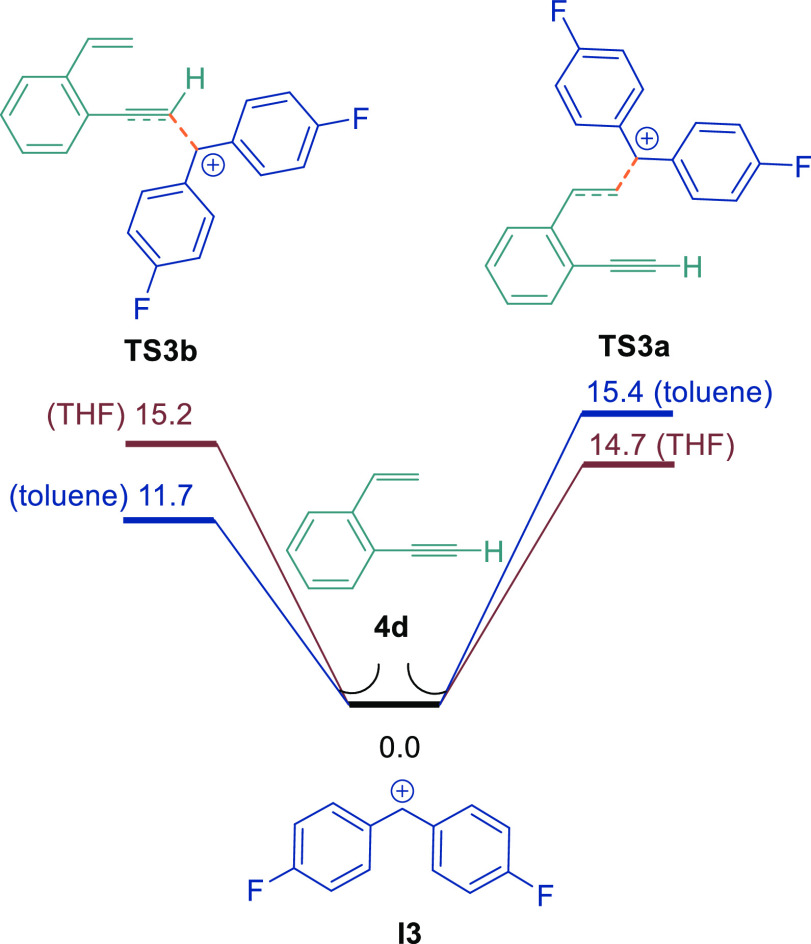
DFT calculations of the site selectivity of I3 with 1-ethynyl-2-vinylbenzene 4d, calculated at the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d) level in THF and toluene.
Conclusions
We have demonstrated new reactivities of FLPs in the functionalization of terminal alkynes through Csp3–Csp coupling reactions with aryl esters. DFT studies found that a diamagnetic pathway was most likely, although a low-energy single-electron pathway could operate to some extent. In particular, DFT studies indicate that the combination of the Mes3P/diaryl methylene cation led to three species of similar energy in solution: the FLP (I3), the Lewis acid–base adduct (I4), and the frustrated radical pair (I5). According to the Curtin–Hammett principle, the reaction proceeds predominantly via TS5 from rapidly equilibrating I3, I4, I5, and I6. These rapidly equilibrating species in solution are supported by the observation of radical species of varying stabilities and lifetimes in the reaction mixture. Thus, radical species are formed in the reaction but are not making a substantial contribution on the reaction pathway to the product, with the possible exception of arylacetylenes with strongly electron-withdrawing (e.g., p-NO2, p-CF3) substituents. These observations will be of importance when designing future reactions that can switch between one- and two-electron pathways depending upon the substrate. Moreover, we observed high site selectivity when ethynyl vinylbenzene substrates were employed in these reactions. 1-Ethynyl-4-vinylbenzene substrates reacted only at the alkyne site, but 1-ethynyl-2-vinylbenzene substrates showed high selectivities depending upon the polarity of the solvent. For 1-ethynyl-2-vinylbenzene in THF, Csp3–Csp coupling was observed, resulting in alkene functionalization, whereas in toluene or TFT exclusive Csp3–Csp coupling and alkyne functionalization resulted. The contrasting selectivity was explained by DFT and computed transition states in the differing solvents. FLP-mediated C–C bond-forming reactions are still relatively new, but there is no doubt that advances will continue to be made in this area. This reported methodology can be utilized to generate compounds that can be subsequently employed for the synthesis of useful novel natural products where metal-free synthesis is highly desirable for avoiding metal toxicities.15,33
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c01622.
Detailed experimental procedures, NMR spectra, EPR data, kinetic data, and DFT data (PDF)
Notes
A.D., K.S., and R.L.M. acknowledge the EPSRC for an Early Career Fellowship for funding (EP/R026912/1). A.D., E.R., and R.L.M thank the Leverhulme Trust for a research project grant (RPG-2020-016). A.A., B.F.Y., and R.B. thank the Australian Research Council (ARC) for project funding (DP180100904) and the Australian National Computational Infrastructure and the University of Tasmania for the generous allocation of computing time. Information about the data that underpins the results presented in this article, including how to access them, can be found in the Cardiff University data catalogue. This can be found at http://doi.org/10.17035/d.2021.0130087985.
Notes
The authors declare no competing financial interest.