 Selective Anion Binding Drives the Formation of
AgI8L6 and AgI12L6
Six-Stranded Helicates
Selective Anion Binding Drives the Formation of
AgI8L6 and AgI12L6
Six-Stranded Helicates
Journal of the American Chemical Society


- Altmetric
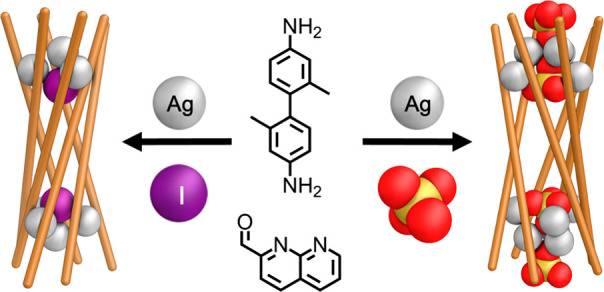
Here we describe the formation of an unexpected and unique family of hollow six-stranded helicates. The formation of these structures depends on the coordinative flexibility of silver and the 2-formyl-1,8-napthyridine subcomponent. Crystal structures show that these assemblies are held together by Ag4I, Ag4Br, or Ag6(SO4)2 clusters, where the templating anion plays an integral structure-defining role. Prior to the addition of the anionic template, no six-stranded helicate was observed to form, with the system instead consisting of a dynamic mixture of triple helicate and tetrahedron. Six-stranded helicate formation was highly sensitive to the structure of the ligand, with minor modifications inhibiting its formation. This work provides an unusual example of mutual stabilization between metal clusters and a self-assembled metal–organic cage. The selective preparation of this anisotropic host demonstrates new modes of guiding selective self-assembly using silver(I), whose many stable coordination geometries render design difficult.
Self-assembly can produce complex metal–organic architectures from simple starting materials.1−5 Such structures have been the subject of intense recent exploration, with applications spanning guest binding, stabilization of reactive species, biomolecular interactions, and chemical purification.6−9 These applications often depend on binding a target in the pseudospherical cavity of a metal–organic cage. These isotropic cavities can bind roughly spherical guests or guest agglomerates10−13 but are ill-adapted to bind asymmetric and anisotropic guests. The introduction of flexible organic ligands14−16 or metal coordination spheres17−20 has led to the formation of new metal–organic cages, with nonspherical internal cavities, partially alleviating these limitations.21−25 Silver(I), in combination with dipyridyl peptidic linkers, has recently been shown to generate a wealth of complex knotted architectures via self-assembly.26−28 The strategy of incorporating a guest of interest into the architecture formed, as a template29−31 or other structural element,32,33 can enhance selectivity and sensitivity in guest binding.34−36 Furthermore, if the guest is anionic,37−39 the diverse coordination chemistry of anions can be used to effect the selective recognition40−42 of targeted anions.43
We hypothesized that the flexible coordination sphere of silver(I) ions,44−49 in combination with organic ligands that assemble in situ around these metal-ion templates, would provide access to new structure types that bind anions as structural elements. Zhao and co-workers have previously shown that nitrogen containing macrocycles can stabilize atomically precise silver clusters with defined geometries, supporting this hypothesis.50,51
Here we describe the formation of a family of complex six-stranded silver helicates upon the addition of three anions: iodide, bromide, and sulfate. This family comprises two novel structure types, with sulfate generating a structure distinct from those templated by halides. Key structural elements within these architectures are unique silver(I)-anion clusters,50,51 whose geometries are molded by the central anions, which in turn are held in an unusual, polarized, environment.
Building on the discovery that silver(I) assembles with 2-formyl-1,8-naphthyridine (1),52 a tritopic subcomponent, and anionic templates to form a trigonal prism with disilver vertices,17 we investigated the use of linear ditopic anilines in place of triangular ones. Initial experiments, involving the mixture of benzidine (2) together with 1, various silver salts, and prospective guests in acetonitrile (Figure 1a), gave in all cases an intractable gel (SI Section 8).

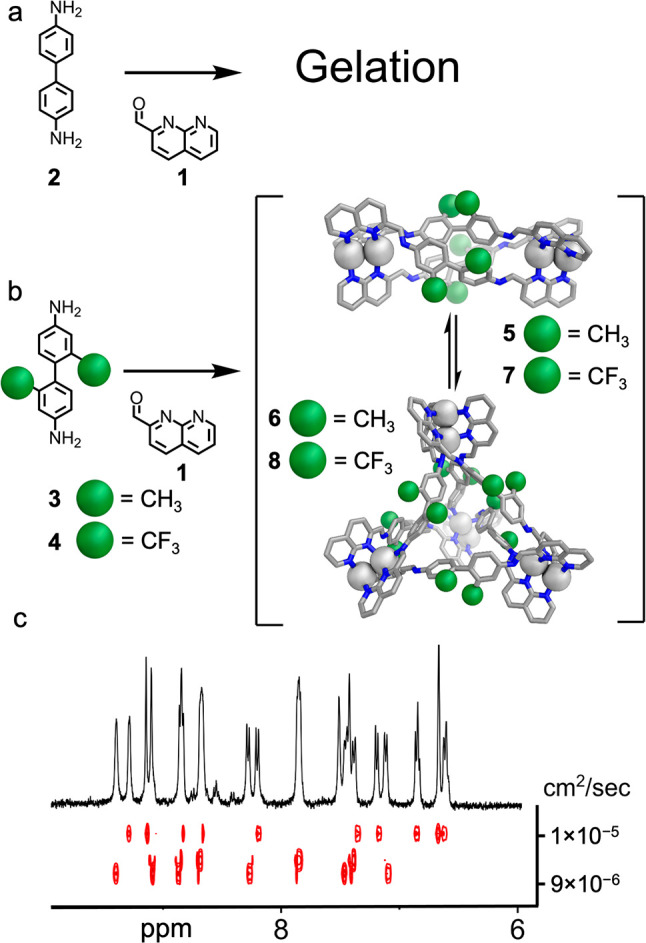
Self-assembly of Ag4L3 and Ag8L6 architectures. Conditions: (a) AgNTf2 (2 equiv), 2 (1 equiv), 1 (2 equiv), d3-MeCN, 5 min; (b) AgNTf2 (2 equiv), 3 or 4 (1 equiv), 1 (2 equiv), d3-MeCN, 5 min. Structures of 5 and 6 are MM3-optimized models. (c) DOSY NMR of 5 and 6.
Reasoning that increasing steric hindrance and widening the torsion angle between the phenylene groups of the dianiline could lead to a different outcome,53 we explored the self-assembly of 2,2′-dimethyl-[1,1′-biphenyl]-4,4′-diamine (3) with 1 in acetonitrile, and observed the formation of discrete species with various silver(I) salts (Figure 1b). With silver perchlorate, we observed a 1:1 ratio of integrals between two species (Figure 1c). Diffusion ordered spectroscopy (DOSY) NMR revealed that one had a significantly larger diffusion coefficient (Figure 1c). Mass spectrometry indicated that the smaller species had Ag4L3 composition, with the larger species corresponding to Ag8L6 (Figures S72 and S75). Approximately 400 attempts to grow crystals of these species failed.
The observation of well-defined bands of peaks in the DOSY spectrum is consistent with the formation of discrete species, as opposed to poorly defined oligomers in solution.54,55 We modeled potential structures for the Ag8L6 architecture and found that a tetrahedral geometry was preferred by 300–400 kcal mol–1 (SI Section 9).56 Although we cannot definitively assign the product structures without crystallographic data, we infer that the two species are likely to be Ag4L3 helicate 5 and Ag8L6 tetrahedron 6, consistent with previously reported systems,57 our modeling studies, and the solution data (SI Section 4.4). Investigations of host–guest behavior showed binding to a range of anionic and organic guests, with some altering the 5:6 equilibrium (SI Section 7).58,59 When dianiline 4 was used in place of 3 we observed similar results (Figure 1b and SI Section 10).
Having extensively screened potential guest species, we next turned to the addition of halides to these silver(I) based assemblies. We had initially avoided the use of halides, anticipating precipitation of silver halide species (the solubility product of AgI is 10–14.5 in acetonitrile).60 However, upon addition of TBA iodide, a new species, 9, immediately formed and, to our surprise, no precipitate was observed.
Characteristic 1H NMR signals were observed for 9 at 6 ppm, ca. 1 ppm upfield of any signals of 5 or 6 (Figure 1c). Furthermore, a twofold desymmetrization was observed, with two 1H NMR signals observed for each proton environment in free ligand (Figure S1). DOSY spectroscopy gave results consistent with the formation of a single species (Figure S8). Mass spectrometry confirmed that a Ag8L6I2 architecture had been formed (Figures S69 and S77).61
The X-ray crystal structure of 9 revealed its highly unusual six-stranded helicate structure (Figure 3a,b), which is capped at each end by a Ag4I cluster consisting of a Ag3 triangle capped by an apical Ag on the outside and iodide on the inside (Figure 3e). The six ligand strands bridge two such Ag4I clusters, grouped into three pairs of ligands that show aromatic stacking interactions between naphthyridine moieties, with distances of 3.1–3.7 Å between stacked rings.
Atypical coordination environments for the Ag centers were observed in 9. One arm of each ligand coordinates via all three available nitrogen donors, and the other via only a single inner naphythridine nitrogen. This differentiation leads to the twofold desymmetrization seen in the 1H NMR spectrum
The presence of 12 uncoordinated nitrogen donors within 9 violates the principle of maximal coordinative saturation, which has often, and successfully, been used to predict the product of metal–organic self-assembly processes.62 The absence of coordinative stabilization may be a consequence of the nonchelating coordination vectors of 1, which precluded the formation of simple structures. The lack of coordinative saturation is compensated for by the extensive aromatic stacking seen in the crystal structure of 9.63
Silver–silver separations were 2.96–3.00 Å between silver atoms bridged by a single naphthyridine moiety, greater than those observed in simpler mononuclear naphthyridine-bridged silver complexes.63 The iodide ion coordinated to all four Ag ions in the cluster, with Ag–I separations of 2.79–2.88 Å, consistent with previous reports of Ag4I clusters.50,51
Having determined the structure of 9, we investigated whether alternative anions might lead to the generation of further examples of this new structure type. Addition of tetramethylammonium sulfate to a mixture of 1, 3, and silver triflimide brought about conversion to an alternate species, 10, as the uniquely observed product (Figure 2). This product again showed twofold desymmetrization in the 1H NMR (Figure S9) and a single species by DOSY NMR (Figure S14). We initially anticipated that a structure analogous to 9 would be formed, with Ag8L6(SO4)2 stoichiometry, based upon similarities between 1H NMR spectra (Figure S9). However, mass spectrometry indicated that instead a Ag12L6(SO4)4 species formed (Figures S70 and S78). Six-stranded helicate formation was confirmed by single-crystal X-ray diffraction (Figure 3c,d). The organic portion of the structure was similar to 9, yet the silver clusters at the ends of both assemblies are dramatically different. Instead of the Ag4I clusters of 9, the vertices of 10 consist of Ag6(SO4)2 clusters composed of inner and outer Ag3 triangles. The externally facing sulfate coordinates to the outer triangle of silver ions via a single, triply coordinated, oxygen atom.64 The coordination of this sulfate is reinforced by nonclassical hydrogen bonding from three naphthyridine CH groups (CH···O distances 2.40–2.43 Å), stabilizing the assembly (Figure 3f).65 Each silver ion of this outer triangle is also coordinated by the internal sulfate via a single, triply coordinated oxygen. The interior sulfate additionally coordinates to the internal, more widely spaced, triangle of silver ions. The two Ag triangles form pairs of silver ions in close proximity, with each bridged by two naphthyridine moieties. The sulfur atoms of the internal anions are 11.58 Å apart, farther than the iodide anions in 9 (10.47 Å), and show nonclassical hydrogen bonds (CH···O distances 2.58–2.69 Å) to internally facing CH groups (Figure 3g). Ligand coordination again shows pairwise alternation, here between three and two coordinating nitrogen atoms per ligand arm. The uncoordinated donor atoms were again imine nitrogens
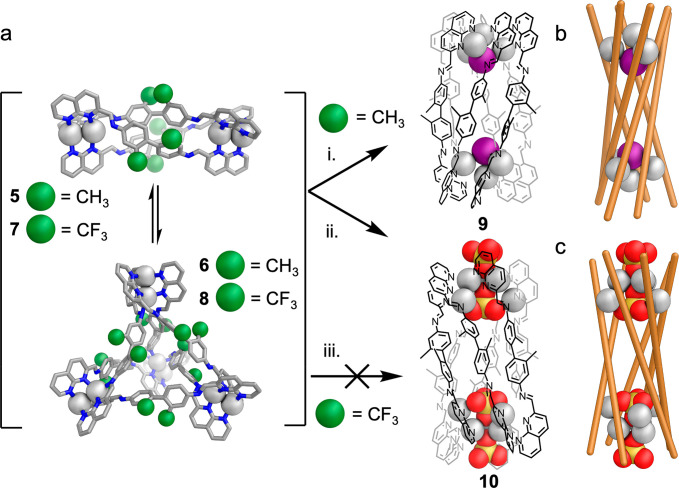
(a) Synthesis of six-stranded helicates 9 and 10, formed only during self-assembly from dianiline 3. (i) Tetrabutylammonium iodide (0.34 equiv), 5 min; (ii) tetramethylammonium sulfate (1.0 equiv), 6 h. Structures of 5 and 6 are MM3 optimized models, and those of 9 and 10 are based on crystallographic data (vide infra). Simplified representation of six-stranded helicate (b) 9 and (c) 10.
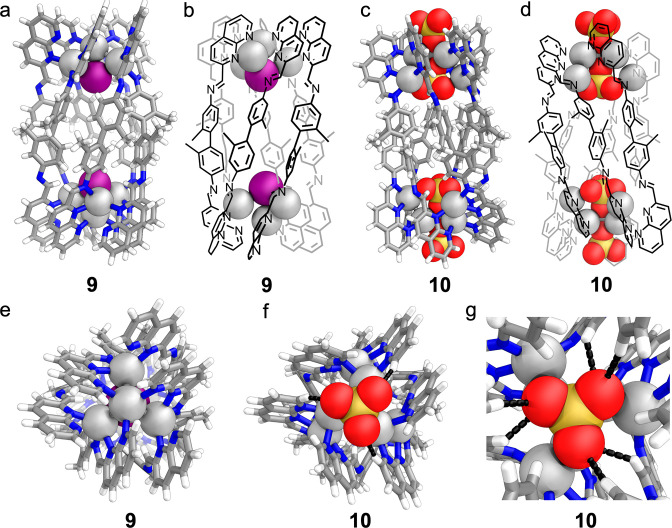
(a) X-ray crystal structure of 9; (b) schematic view of 9. (c) X-ray crystal structure of 10; (d) schematic view of 10. (e) End-on view of crystal structure of 9 showing cluster geometry. (f) End-on view of crystal structure of 10 showing the silver cluster and nonclassical hydrogen bonds to the exterior sulfate. (g) View from within the crystal structure of 10, showing nonclassical hydrogen bonds to the internal sulfate.
We next investigated whether other anions could template structures similar to 9 and 10. Among the 38 anions tested (SI Sections 6.7 and 6.8), only bromide proved able to efficiently template a six-stranded helicate (11). The 1H NMR spectrum of 11 again exhibited a twofold desymmetrization, and a single species was observed by DOSY spectroscopy, with a hydrodynamic radius of 11.9 Å, similar to the cases of 9 and 10 (Figures S8, S15, and S22). Attempts to grow crystals suitable for X-ray diffraction proved unsuccessful. However, we inferred the Ag8L6Br2 structure of 11 to be an analogue of 9 by comparing the 1H NMR, COSY, and HSQC spectra of 9–11. The spectra of 9 and 11 were clearly similar, whereas that of 10 was notably different (Figure 4a and SI Section 5).
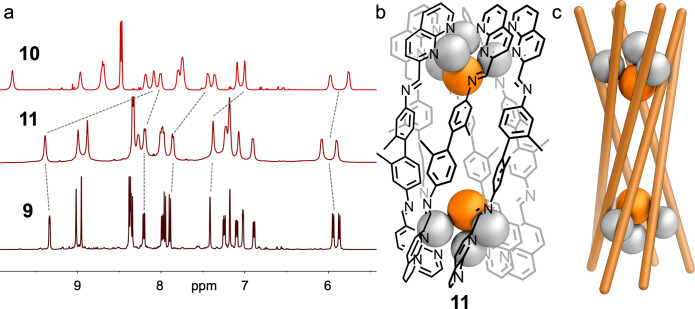
(a) Comparison of 1H NMR spectra of 10 (top), 11 (middle), and 9 (bottom), showing the similarity between the spectra of 9 and 11. Simplified (b) schematic and (c) cartoon views of six-stranded helicate 11.
We then probed further the selectivity of the assembly process. Silver tetrafluoroborate, hexafluorophosphate, perchlorate, and triflate all furnished six-stranded helicates adopting the framework of 9 when combined with 1, 3, and potassium iodide (Figures S38 and S39). Titration of TBA bromide into a mixture of 5 and 6 revealed no intermediate species (i.e., from binding a single bromide). Instead, formation of 11 (containing two bromide anions) was seen immediately, in the continued presence of 5 and 6 (Figures S42 and S46), suggesting that the six-stranded helicate assembled cooperatively (SI Sections 6.3 and 6.6). Using 2 or 4 in place of 3 led to immediate gelation (for 2) or shifts in the equilibrium of 7 and 8 (for 4, Figures S55 and S68).
These results highlight the extent to which the subcomponent self-assembly of metal–organic architectures may depend critically upon subtle variations in subcomponent structure. The lack of methyl groups on 2 favored polymerization over the assembly of discrete structures. The subtle steric and electronic differences between the methyl groups of 3 and the trifluoromethyl groups of 4 disfavored, in the latter case, the formation of six-stranded helicates analogous to 9–11. We hypothesize this sensitivity to be due to the slightly weaker ligand field in the case of ligands incorporating 4, which disfavors structures that incorporate the more highly cationic silver clusters incorporated into the new structure types 9–11.
This work describes the development of a system of novel six-stranded helicates, which assemble around atomically precise silver clusters. Specific anionic templates, in turn, serve to shape these clusters, such that the identity of the anion dictates the architecture observed. The ability of 2-formyl-1,8-napthyridine to bridge silver ions enables these complex structures to form from simple subcomponents. These new assemblies are sensitive to the precise nature of the ligand chosen and are selective for the templates employed, with potential applications in sensing specific analytes.
The ability to use atomically precise clusters in place of mono- or dimetallic vertices in metal–organic cages has the potential to generate a vastly increased diversity of architectures, as we continue to uncover the principles underpinning silver–naphthyridine self-assembly. Future work will focus on exploring the photophysical properties of these novel clusters66 and on expanding the range of architectures formed by the interplay of anion templation, ligand design, and coordinational flexibility to generate increased structural diversity.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c11905.
Notes
The authors declare no competing financial interest.
Acknowledgments
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1) and the European Research Council (695009). We thank the University of Cambridge Mass Spectrometry Service Centre for high-resolution mass spectrometry and Diamond Light Source (UK) for synchrotron beamtime on I19 (CY21497). C.T.M. thanks the Leverhulme Trust and the Isaac Newton Trust, and Sidney Sussex College, Cambridge, for Fellowship support.
References
Although the coordinative flexibility shown by the napthyridine–silver system limits the degree of certainty of these modeling results, the large difference in energy between the tetrahedral architecture and alternate structures lends credence to the assignment of the Ag8L6 structure as a tetrahedron.
Mass spectrometry of these silver complexes is challenging, presumably due to the dynamic nature of the naphthyridine–silver interactions. We see extensive fragmentation of all complexes under even mild conditions. By tuning ionization conditions and through choice of counterion, we were able to gather data on these architectures in both LRMS and HRMS. We found that using hexafluorophosphate as the counterion was particularly effective for obtaining good quality mass spectra.
A minor isomer was also resolved in the crystallographic data, whereby an exterior sulfate coordinates via three oxygen atoms instead. Please see SI Section 12 for further details.
These silver–naphthyridine systems proved to be extremely stable to light, which was unexpected. Samples could be left exposed to ambient light for 2–3 months with no sign of decomposition by NMR, or precipitation.