 Genome-in-a-Box:
Building a Chromosome from the Bottom
Up
Genome-in-a-Box:
Building a Chromosome from the Bottom
Up
ACS Nano


- Altmetric
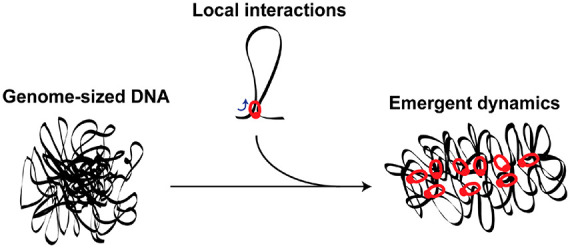
Chromosome structure and dynamics are essential for life, as the way that our genomes are spatially organized within cells is crucial for gene expression, differentiation, and genome transfer to daughter cells. There is a wide variety of methods available to study chromosomes, ranging from live-cell studies to single-molecule biophysics, which we briefly review. While these technologies have yielded a wealth of data, such studies still leave a significant gap between top-down experiments on live cells and bottom-up in vitro single-molecule studies of DNA–protein interactions. Here, we introduce “genome-in-a-box” (GenBox) as an alternative in vitro approach to build and study chromosomes, which bridges this gap. The concept is to assemble a chromosome from the bottom up by taking deproteinated genome-sized DNA isolated from live cells and subsequently add purified DNA-organizing elements, followed by encapsulation in cell-sized containers using microfluidics. Grounded in the rationale of synthetic cell research, the approach would enable to experimentally study emergent effects at the global genome level that arise from the collective action of local DNA-structuring elements. We review the various DNA-structuring elements present in nature, from nucleoid-associated proteins and SMC complexes to phase separation and macromolecular crowders. Finally, we discuss how GenBox can contribute to several open questions on chromosome structure and dynamics.
Ever since Watson and Crick discovered that the innate double-helix structure of DNA was key to its hereditary function,1 a major question has been how the physical structure of the genome underlies its biological function. Historically, the study of chromosomes started at the phenomenological level already in the 19th century, when Flemming reported on the changing shape of chromosomes across the phases of the cell cycle from interphase to mitosis.2 Significant progress was made in the second half of the 20th century when the molecular biology revolution opened access to studying the many nanoscopic elements that underlie chromosomal structure–a development that has continued to the present day. The past decade, in particular, rapidly expanded our knowledge of how the genetic material is physically organized within the cells of the various kingdoms of life, yielding a string of notable discoveries on the interplay between function, structure, and dynamics of chromosomes. Breakthroughs were, for example, the structural mapping of the genomes using chromosome conformation capture (Hi-C and related) techniques,3,4 the capability of structural maintenance of chromosomes (SMC) protein complexes to extrude loops of DNA as demonstrated using single-molecule fluorescence assays,5−9 how the properties of the cytoplasm, nucleoplasm, and confinement influence the dynamics and structure of chromosomes across species,10−19 or the discovery of the importance of phase separation in various aspects of chromosome organization,20−25 from transcriptional condensates26−33 to heterochromatin formation.34−36 From this brief list, it is already apparent that chromosome organization spans a multitude of scales from single molecules to full chromosomes.
Eukaryotes and prokaryotes organize their genomes differently, storing a large 107–1011 basepair (bp) eukaryotic genome37 in multiple chromosomes inside a nucleus (Figure 1a) versus packaging a smaller 105–107 bp prokaryotic genome37 in one chromosome, also called the nucleoid, that is freely floating within the cell cytosol (Figure 1b). Yet, the basic genetic material, the double-helix DNA polymer, is the same, and it is becoming clear that there are many homologies indicating similar building principles across the various kingdoms of life. Indeed, in this review, we will stress the similarities between the organization of eukaryotic and prokaryotic organisms.

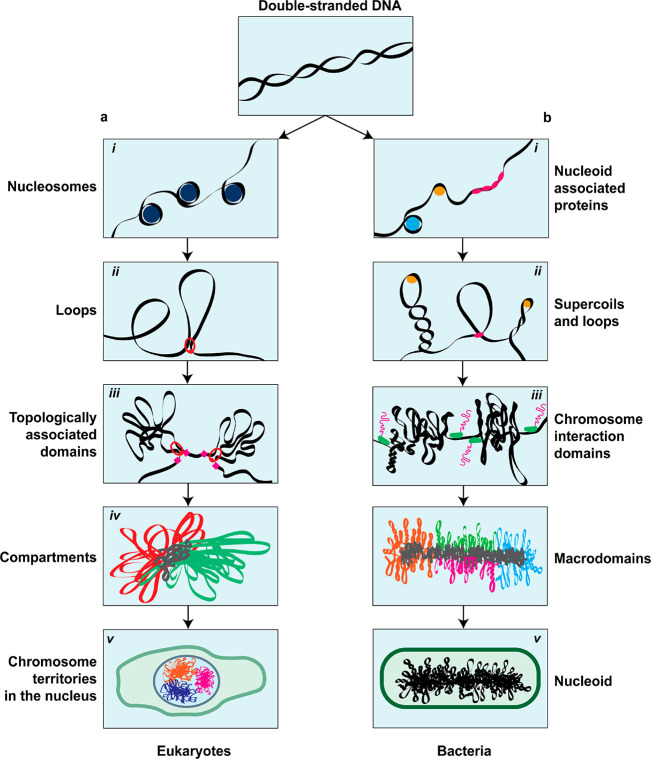
The hierarchical chromosome organization in eukaryotes and bacteria. Double-stranded DNA is the basic component of all chromosomes in both (a) eukaryotes and (b) prokaryotes. (a, i) DNA in eukaryotes is wrapped around histones into nucleosomes, forming a beads-on-a-string structure. (a, ii) Loops are formed through passive bridging or active loop extrusion. (a, iii) TADs are large-scale structures that have increased contact frequency among their DNA loci. (a, iv) Epigenetic markers define if parts of the genome are either transcriptionally active (euchromatin) or repressed (heterochromatin), which are spatially organized in A- and B-compartments, respectively. (a, v) Within the eukaryotic nucleus, chromosomes each occupy their own ‘territory’ that is segregated from the other chromosomes. (b, i) In bacteria, the local structure of the DNA is modulated by NAPs. (b, ii) Most DNA in bacteria is negatively supercoiled, forming plectonemes. Additionally, bridging proteins and SMCs form loop-like structures. (b, iii) Actively transcribed long genes form boundaries for plectonemes, demarcating CIDs. (b, iv) On a larger scale, the circular bacterial genome is organized in macrodomains. (b, v) The bacterial chromosome, called the nucleoid, is embedded in the cytosol and confined by the cell boundary.
At the most basic level (Figure 1), nanometer-sized proteins such as histones38 or bacterial nucleoid-associated proteins39,40 bind the DNA, where they locally modulate the structure and mechanical properties of the DNA, thereby establishing a “beads-on-a-string” conformation. These chromatin fibers are further organized in loop-like structures that are formed either through the action of protein complexes5−9,41,42 or via supercoiling43−45 by twisting the DNA about its axis. Larger-scale levels of organization are characterized by the amount of interactions or contact frequencies that DNA loci have with each other. At the scale of ∼300 nm or 105–106 bp, topologically associated domains (TADs) have been identified in eukaryotes,3,46 while their counterparts in bacteria are called chromosome interaction domains (CIDs),47 which are at the scale of 104–105 bp. Beyond the level of TADs/CIDs, bacteria have macrodomains,48,49 while in eukaryotes alternating chromosomal regions (compartmental domains) are segregated into two types of compartments that feature either relatively high or low gene-expression levels, and which are collectively called A-compartments (euchromatin) and B-compartments (heterochromatin), respectively.3,50 Finally, in the nucleus, individual chromosomes do not mix, but each occupy distinct locations called chromosome territories, albeit with a limited and transcription-dependent overlap between them.51−53 The cell cycle, and in particular cell division, is associated with major rearrangements of the chromosomal structure. During interphase in eukaryotes, chromosomes are geared toward accessibility and gene expression, whereas in mitosis, the structure is strongly compacted into a bottlebrush structure for faithful transmission of the genetic material to daughter cells.54 Bacteria, by contrast, do not possess such distinct mitotic and interphase chromosome structures. Nevertheless, they also regulate the spatial segregation of replicated chromosomes before cell division.45 Despite this broad spectrum of different phenomenological aspects in the organization of genomes, it increasingly appears possible to explain major characteristics of chromosome organization by a limited number of overarching physical principles,23,25 such as polymer physics, DNA looping, and phase separation.
In this review, we first make a concise survey of various experimental techniques to study chromosome organization and the type of information that these techniques yield about DNA-organizing elements and their local mechanisms. Then we describe an alternative experimental approach, coined ‘genome-in-a-box’ (GenBox), which is an in vitro method for studying genome-sized DNA to which purified DNA-organizing elements can be added. Subsequently, we provide an overview of how various such ‘chromosome building blocks’ contribute to chromosomal organization. Finally, we elaborate in what manner GenBox can contribute to several relevant scientific questions in the field.
Complementary Approaches to Study Chromosome Organization
A wide range of methods is available for studying chromosome organization. Broadly speaking, one can use in vivo studies in either live or fixed cells or in vitro single-molecule biophysical methods. These approaches provide complementary information about chromosome organization and the various DNA-organizing elements.
Methods that explore chromosomes in cells fall into two broad categories: fluorescence-based imaging and methods involving sequencing and immunoprecipitation. Fluorescence-based methods55,56 require a fluorescent reporter for visualization. These reporters can be nonspecifically targeted to the DNA on a global level (e.g., a DNA dye) or locally in a sequence-specific manner via hybridization of a fluorescent oligonucleotide to a complementary sequence (e.g., Oligo-PAINT FISH-probes57,58) or via the binding of a fluorescently labeled protein to its specific DNA-binding site. Examples of the latter include CRISPR-dCas959,60 that binds to a site defined by the guide RNA, operators binding to arrays of repressor sites (FROS arrays),61,62 or ParB proteins binding and oligomerizing near parS sites.63,64 Using these labeling techniques and (super-resolution) microscopy, structural and dynamic data can be collected across a wide range of time and length scales.12,46,65
Methods based on sequencing and immunoprecipitation make it possible to figure out three types of information for each locus on a chromosome: (i) the average proximity of a particular DNA locus to other loci, resulting in a contact-frequency map66,67 (e.g., Hi-C and related techniques); (ii) what proteins are bound or not bound to a specific locus, revealing a map of either protein–DNA interactions68 (e.g., ChIP-seq or DamID) or DNA-accessibility69 (e.g., MNAse-seq or ATAC-seq); and (iii) a combination of these two, in order to, for example, show what proteins mediate a particular long-range interaction70,71 (e.g., Hi-ChIP or ChIA-PET). The aforementioned methods generally result in population-averaged data, making it difficult to determine how the presence or absence of a feature on a interaction map might correspond to the situation inside a single cell.72,73 To counter this, single-cell or single-molecule alternatives have been developed, such as single-cell Hi-C74 for loci contact mapping, single-molecule ATAC-seq (SMAC-seq75) for DNA-accessibility mapping, or single-cell DamID76 to map protein–DNA interactions. Furthermore, these methods can be combined with transcriptome profiling, in order to get insight in the relationship between local genome structure and gene expression,77,78 for example, scDAM&T-seq79 combines single-cell DamID with mRNA sequencing.
A diverse array of single-molecule biophysics techniques can be used to study DNA and its binding proteins. In DNA curtains and other visualization assays,80,81 long DNA molecules (up to 50 kbp) are attached to a surface in a flow cell, which allows time-resolved fluorescence imaging of the stretched DNA and the action of single proteins thereupon. Atomic force microscopy (AFM)82,83 provides a label-free scanning probe technique, resulting in a topographic map of the (typically dried) sample at nanometer resolution. Additionally, AFM can be used for dynamics since it is able to image at video rates in liquid, which enables to observe, at the single-molecule level, conformational changes of a protein while it interacts with DNA. Transmission electron microscopy (TEM) uses electrons to image a fixed sample with superb angstrom-level resolution, but it needs a vacuum environment and the imaging contrast depends on the use of staining agents and sample thickness. Cryo-electron microscopy84−87 is best suited for biological samples, as the biomolecule of interest is embedded inside a thin layer of amorphous ice, yielding three-dimensional structures at subnanometer resolution. Optical FRET assays use the principle of Förster energy resonance transfer,88−90 in which energy is transferred between two fluorophores, depending on the distance between the molecules. Upon site-specific fluorescent labeling, FRET can be used to measure time-resolved nanometer-scale conformational changes of the protein and the DNA. In magnetic tweezers,91 a DNA molecule is attached between a surface and a bead, of which the position and rotation can be manipulated by a magnet. This allows to get information about the force (at subpiconewton resolution) or torque that DNA-structuring proteins exert on the DNA. Optical tweezers92,93 use a focused laser beam to trap one or more beads, to which biomolecules such as DNA are attached. Manipulation of the beads enables force spectroscopy on single molecules as well as complicated topological perturbations, for example, to enable the construction of complex protein bridges between two DNA molecules. Notably, it is possible to combine optical tweezers with FRET, confocal fluorescence microscopy, and super-resolution microscopy, making it possible to observe conformational changes, binding kinetics, and localization of fluorescently labeled proteins to DNA as a function of applied force.
Chromosomes have also been studied extensively in silico.94−98 Modeling a chromosome and the effects of DNA-structuring proteins starts with modeling DNA itself, for which there is a range of parameters that can be tweaked: the total polymer length, the persistence length, attractive or repulsive interactions between DNA monomers on either a global scale or locally between specific monomers, the composition and quality of the solvent and surrounding medium, the level of supercoiling dictated by the amount of twist and writhe, the topology of the DNA (linear, circular, knotted), and the confinement volume and geometry. Due to the relative ease of scanning these parameters individually or in various combinations, computer simulations have been a very fertile ground for studying DNA organization. An intriguing early example was the finding that two genome-sized polymers spontaneously demix and spatially segregate inside a cylindrical confinement, related to the entropy of (de)mixing of chromosomes19 (Figure 2a). More recently, by using simple principles of multivalent interactions and bridging by DNA-binding proteins, simulations of phase separation showed clusters relevant for chromosome structure99 (Figure 2b). Furthermore, models have been built for eukaryotic chromosomes, showing, for example, that loop-extruding elements acting on DNA can disentangle newly replicated DNA into structures that closely resemble mitotic chromosomes4,100 (Figure 2c). Loop-extrusion polymer-simulation models can also recapitulate the TAD structures found in interphase chromosomes.101,102
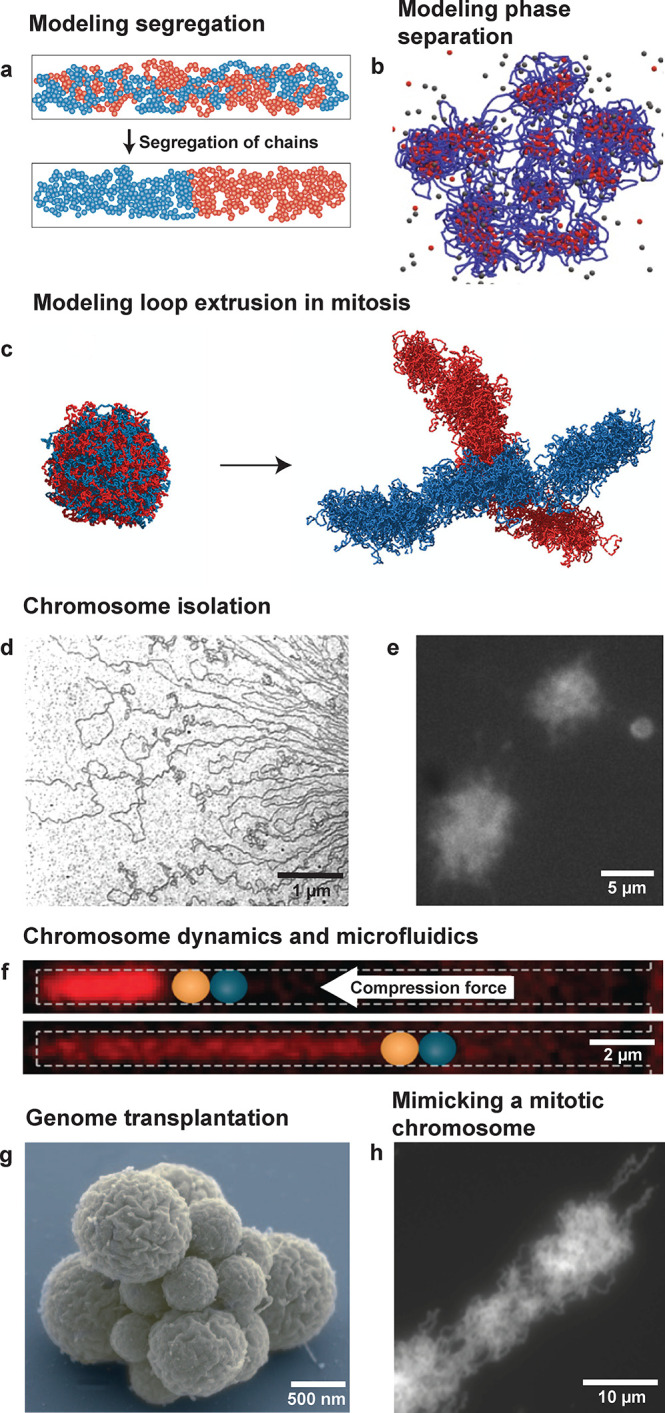
Examples of previous in silico and in vitro research on genome-sized DNA. (a) Confinement can induce the entropic demixing of two long polymers. Counterintuitively, the segregated state has a higher entropy than the mixed state. Adapted with permission from ref (168). Copyright 2010 Springer Nature. (b) DNA-binding proteins that bridge DNA can lead to phase separation into clusters. Adapted with permission from ref (96). Copyright 2020 Springer Nature. (c) SMC loop extruders can segregate a replicated random polymer into an object resembling a mitotic chromosome. Adapted with permission from ref (100). Copyright 2016 Goloborodko et al. (d) Electron microscopy image of an E. coli chromosome, showing supercoiled plectonemes. Adapted with permission from ref (115). Copyright 1976 Springer Nature. (e) Fluorescence image of isolated E. coli chromosomes in solution. Adapted with permission from ref (169). Copyright 2012 Elsevier. (f) An E. coli chromosome is compacted by a piston formed by an optical tweezer bead (blue) inside a micron-sized channel. Adapted with permission from ref (117). Copyright 2012 National Academy of Sciences. (g) A synthetic genome can be transplanted into a host cell, which leads to the creation of a synthetic cell JCVI-syn3.0, shown here. Adapted with permission from ref (119). Copyright 2016 The American Association for the Advancement of Science. (h) Frog-sperm chromatin can be combined with six purified protein complexes to yield structures similar to mitotic chromosomes. Adapted with permission from ref (120). Copyright 2015 Springer Nature.
These approaches each have their advantages and drawbacks. The main strength of in vivo live-cell studies is, obviously, that they inherently examine chromosomes within the natural context of the genomic polymer, the living cell. Their major downside is the vast complexity of the inner environment of cells with their multitude of simultaneously interacting biomolecular components. This makes it challenging to provide clear cause–effect relations. In vitro biophysics experiments, on the other hand, provide detailed and mechanistic information at the single-molecule level with clear cause–effect relations about specific DNA–protein interactions. However, these experiments are generally performed on short DNA fragments that interact with only one or a few purified proteins near a surface, and as such, they are quite detached from the natural cellular environment. Indeed, the strength of the single-molecule approach is at the same time its weakness, as it does not allow to probe the bigger picture of the combined effect of these DNA-structuring elements on the genome as a whole. In silico experiments are able to study full genomes (by coarse-graining the polymer to a relevant length-scale) with single-parameter control. However, one-to-one corresponding experimental verification of such in silico results is often lacking.
Building a Synthetic Cell from the Bottom Up
In recent years, synthetic biology has gained traction as a third experimental avenue for studying living systems.103 Synthetic cell research deals with the construction of new biological molecules and systems in order to redesign those found in nature, and it does so in one of two approaches. In a top-down approach, synthetic circuits are added to cells, or nonessential elements of living cells are stripped away in an attempt to establish a minimally functional cell. In a bottom-up approach, on the other hand, one tries to compose minimal sets of components that can perform rudimentary functions of living cells. In particular, the aim is to first build modules to establish functional cellular subsystems in isolation, before combining them at a later stage into a synthetic cell. Examples of such cellular modules could be circuits for a machinery for cell division,104,105 transcription–translation for genetic information transfer,106 pattern formation for spatial control,107 and cell–cell communication.108 This approach to synthetic cell research can be called “bottom-up biology”, since its goal is to establish biological function from the bottom up, that is, to construct the essential characteristics of living cells out of a set of well-understood but lifeless components. Notably, various projects have been started across the world that aim at building a synthetic cell.109−112
Although also an in vitro methodology, the bottom-up biology approach significantly exceeds the single-molecule biophysics methodology in multiple ways. First, it literally is scaling up by orders of magnitude, from single proteins to elaborate but controlled protein mixtures and from local molecular-level interactions to collective behavior and their emergent effects. Second, bottom-up synthetic-cell research specifically aims to study the functional subsystems within mimics of the cellular container, for which there is a wide range of possible scaffolds113 (e.g. liposomes, droplets, polymersomes, or microfabricated chambers) and microfluidic technologies104,114 to manipulate them.
Can the bottom-up approach beneficially be applied to study whole chromosomes? In the 1970s, bacterial chromosomes were isolated from cells and prepared for electron microscopy imaging, showing DNA supercoiled loop structures (Figure 2d).115 About two decades ago, Woldringh et al. provided a relatively simple method to isolate bacterial chromosomes from cells for optical microscopy (Figure 2e).116 Jun et al. used this method to study such nucleoids inside microfluidic channels, providing insights into the effects of confinement and macromolecular crowding on DNA organization (Figure 2f).117 Genome transplantation, as developed by Glass et al.,118 made it possible to isolate a chromosome from a cell, remove the DNA-binding proteins, and insert this bare genome into a host cell that had its genome removed. This “rebooted cell” was then able to grow and multiply.118 This approach has been expanded by using a synthetic minimized genome for the purpose of finding a functional minimal version of the original genome (Figure 2g).119 While most of the early efforts involved taking chromosomes out of living cells and simply observing them, Hirano et al. attempted to mimic the construction of mitotic chromosomes in vitro when they combined frog sperm chromatin with six purified protein complexes, leading to structures that, at face value, appeared strikingly similar to mitotic chromatids (Figure 2h).120 This approach was an extension of experiments involving reconstituted chromatin in Xenopus leavis egg extract,121,122 which also included examples of the use of microfluidics to encapsulate the egg extract together with reconstituted chromatin to explore the influence of confinement on the size of the mitotic spindle.123,124
A Genome-in-a-Box
Here, we would like to argue that the time is ripe to embark on an effort to build chromosomes from the bottom up, that is, to establish, in vitro, the full complexity of prokaryotic or eukaryotic chromosomes from basic elements through a systematic hierarchical assembly. We coin this approach ‘genome-in-a-box’ (GenBox). This name derives inspiration from the “particle-in-a-box” models that famously provided basic insights in quantum mechanics, as we hope that GenBox may similarly help to unravel key properties of chromosomal organization. This approach is grounded in the fields of bottom-up biology and synthetic-cell research and extends upon previous research on genome-sized DNA, as described above.
In GenBox (Figure 3), we envision to first isolate chromosomes from cells and strip them of all DNA-binding proteins, resulting in a genome-sized deproteinated DNA substrate, similar to the sample-preparation steps in genome transplantation.118 Subsequently, one can add purified DNA-structuring elements (mostly protein complexes), which can be seen as ‘chromosome building blocks’, with the aim to study their specific effect on the structure and dynamics of the genome-sized DNA. There is a wide range of such building blocks known, for example, SMC protein complexes, topoisomerases, RNA polymerases, crowders, etc., which are reviewed below. Finally, microfluidics and liposome technologies can be used to define a cell/nucleus-sized confinement—the ‘box’ part of the GenBox. Using this scheme, it will be possible to perform in vitro studies of chromosomes in a regime of previously unexplored DNA sizes with great control of the mutual interactions between the various actors. Additionally, it will allow to study chromosome organization in vitro at all its hierarchical levels, with examples ranging from simple DNA-binding proteins inducing various levels of compaction to the influence of loop formation at a global scale and to the interaction between multiple chromosomes in the same confining container as a mimic of chromosome territories. In the spirit of Richard Feynman’s famous saying, “What I cannot create, I do not understand”, GenBox uses the concept of ‘building leads to understanding’ in order to study chromosome organization.
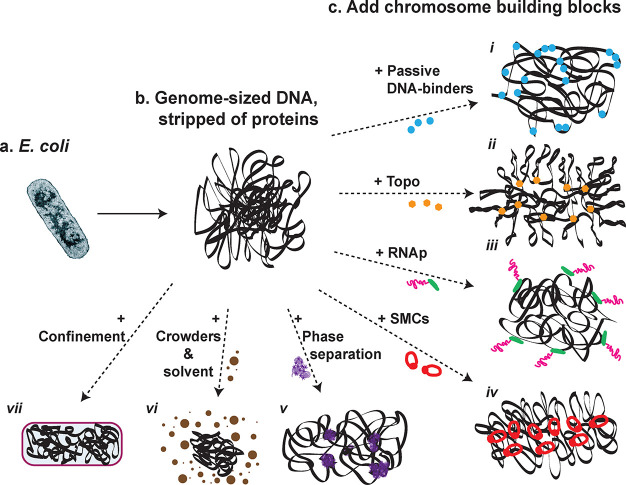
GenBox: recreate genome structure by mixing genome-sized DNA and chromosome building blocks. (a) Chromosomes can be extracted from live cells, for example the circular bacterial chromosome from E. coli. (b) Stripping chromosomes of its DNA-binding proteins results in genome-sized DNA molecules that act as a substrate for subsequent experiments. (c) Next, DNA-structuring elements are added, one at a time. Various categories of such chromosome building blocks are indicated: (i) Passive DNA-binding proteins. (ii) Topoisomerases (Topo) involved in supercoiling and decatenation control. (iii) RNA polymerase (RNAp) responsible for transcription. (iv) SMC proteins that underlie the looping structure of chromosomes. (v) Phase separation that is implicated in transcriptional condensates and the formation of compartments. (vi) Crowders and solvent molecules that modulate compaction of a polymer through entropic depletion forces and solvent–polymer interactions, respectively. (vii) Confinement provided by the cellular or nuclear boundary in cells, which can be mimicked by artificial cell-sized containers.
A key feature in this approach is the use of genome-sized DNA as a substrate. Notably, ‘genome-sized’ is not a very accurate descriptor since genomes from different species vary over 6 orders of magnitude in size, from 0.6 megabasepair (Mycoplasma genitalium125) to 150 gigabasepair (Pieris japonica126). The relevant point, however, is that emergent effects can be expected to come into play in the large-scale DNA organization once the substrate size approaches the ∼ megabasepair range, where, for example, TADs and compartmentalization occur.3,46,50 The source (organism) of the DNA can in principle be freely chosen, as many major features of chromosomal structures occur widely across the domains of life. There are examples, however, where it is desirable to include species-specific sequences on the DNA substrate, because a particular DNA-structuring element needs that sequence to function. For example, CTCF sites (and associated proteins) are crucial in human interphase chromosome organization due to their interaction with cohesin SMCs,3 but these CTCF sites are absent in nonmetazoan eukaryotes and bacteria.
As indicated above, an extensive toolbox of techniques is available to study chromosome structure and dynamics. The most obvious read-out in GenBox experiments would, in first instance, be time-resolved fluorescence imaging that provides dynamic structural information. Global information about the density distribution of the DNA in space and time can be monitored with DNA dyes, and local dynamics of specific spots along the genome can be quantified using sequence-specific fluorescent labels. Fortunately, in vitro experiments allow for much relaxed constraints regarding phototoxicity and choices of fluorophores, in contrast to live-cell imaging. Probing the functional relevance of the GenBox chromosomes will be a next step. As, for example, DNA-binding proteins can lead to structures of varying degrees of compaction, gene accessibility and expression may be influenced. The ability of transcription machinery to transcribe a set of genes can be monitored, for example, by quantitative PCR (qPCR). In parallel to imaging and functional qPCR assays, chromosome conformation capture experiments on these GenBox chromosomes can provide high-resolution information on how a particular chromosome building block affects the contact frequencies among loci.
An Overview of Chromosome Building Blocks
One underlying assumption in this approach is that, to first approximation, chromosome organization can be decomposed into the action of various chromosome building blocks that each have their distinct effect (Figure 4). Below we provide a brief overview of some major chromosome building blocks, which gives a glimpse of the diversity of components involved in chromosome organization.
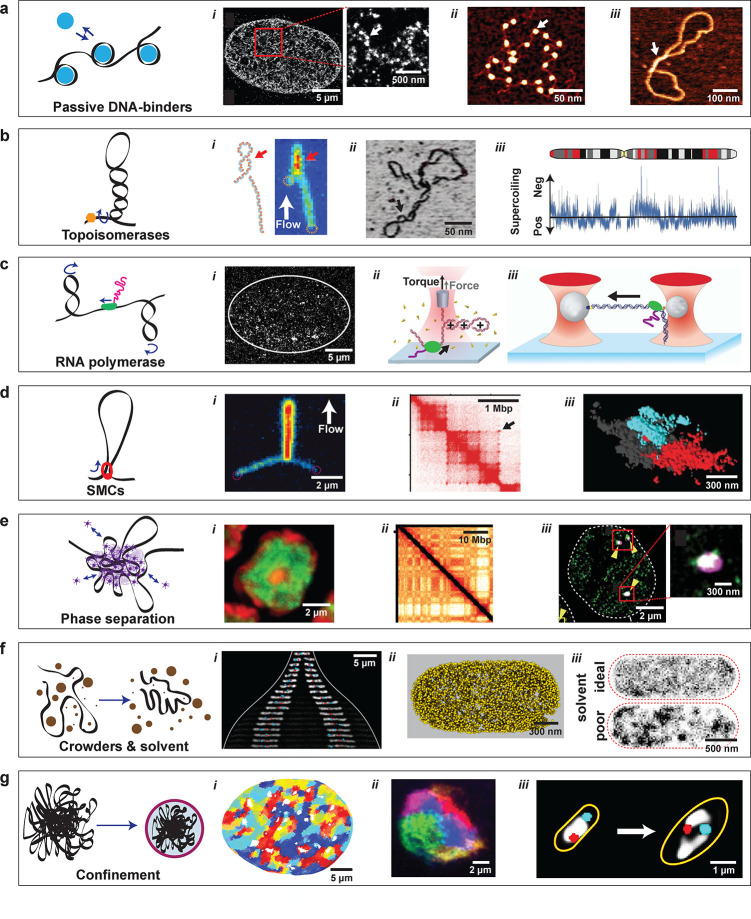
Chromosome building blocks: the elements that constitute chromosome organization. (a) Local DNA-binding proteins bend, wrap, or bridge DNA. (i) Super-resolution image of a eukaryotic nucleus with fluorescently labeled histones. Red box is zoomed in on the right: arrow points to a nucleosome nanodomain (‘nucleosome clutch’) composed of a small number of nucleosomes. Adapted with permission from ref (170). Copyright 2015 Elsevier. (ii) AFM image of DNA with nucleosomes (arrow). Adapted with permission from ref (171). Copyright 2009 Springer Nature. (iii) AFM image of DNA with an H-NS bridge (arrow). Adapted with permission from ref (172). Copyright 2017 The American Society for Biochemistry and Molecular Biology, Inc. (b) Topoisomerases control DNA supercoiling by inducing or relaxing supercoils in the DNA. (i) Optical image and schematic of a plectonemic supercoil on a flow-stretched 20 kbp DNA molecule. Red arrow indicates the plectoneme. Adapted with permission from ref (173). Copyright 2016 American Chemical Society. (ii) AFM image of a supercoiled plasmid. Adapted with permission from ref (174). Copyright 2018 Springer Nature. (iii) Supercoiling density varies between negative and positive along a eukaryotic chromosome. Adapted with permission from ref (175). Copyright 2013 Springer Nature. (c) RNA polymerase (RNAp) transcribes genes in DNA. (i) Super-resolution image of labeled RNAp in a nucleus (white line). The RNAp is found to be nonhomogeneously distributed in small clusters. Adapted with permission from ref (176). Copyright 2013 The American Association for the Advancement of Science. (ii) Optical tweezers with a quartz cylinder can probe both the force and torque exerted by an RNAp (green) acting on a short DNA molecule. Adapted with permission from ref (177). Copyright 2019 National Academy of Sciences. (iii) Optical tweezers can probe the stepping of RNAp (green) along DNA. Adapted with permission from ref (178). Copyright 2006 Royal Society of Chemistry. (d) SMC complexes extrude loops of DNA and are involved in the formation of TADs. (i) Fluorescence image of a flow-stretched DNA molecule, in which an SMC condensin has extruded a loop. Adapted with permission from ref (5). Copyright 2018 The American Association for the Advancement of Science. (ii) A section of a Hi-C contact map, showing TADs (squares) and loops (dots, see arrow). Adapted with permission from ref (155). Copyright 2017 Elsevier. (iii) Super-resolution image of two TAD-like domains (red and cyan) labeled by multiple rounds of FISH Oligopaint. Adapted with permission from ref (46). Copyright 2018 The American Association for the Advancement of Science. (e) Phase separation in chromosomes can occur through multivalent interactions between DNA-binding proteins. (i) Heterochromatin (red) and euchromatin (green) segregate within the nucleus through phase separation. Adapted with permission from ref (34). Copyright 2019 Springer Nature. (ii) A section of a Hi-C map showing a checkerboard pattern indicating that alternating regions of a chromosome interact over large distances through the formation of A and B compartments. Adapted with permission from ref (34). Copyright 2019 Springer Nature. (iii) Super-resolution image of a transcriptional condensate (red boxes) of mediator-coactivator (magenta) and RNAp (green) inside the nucleus (white outline). Adapted with permission from ref (30). Copyright 2018 The American Association for the Advancement of Science. (f) Macromolecular crowders and the solvent quality of the cytosol or nucleoplasm can modulate the compaction of DNA. (i) Crowding influences the expansion and position of two chromosomes (white) within an E. coli cell that expands in size from top to bottom (cell outer edge shown as white line). Adapted with permission from ref (14). Copyright 2019 Elsevier. (ii) Distribution of ribosomes (that act as crowders) in E. coli from cryo-electron tomograms. Adapted with permission from ref (10). Copyright 2020 Xiang et al. (iii) Computer simulation that shows that DNA in a poor solvent (bottom) forms heterogeneous structures, while DNA in an ideal solvent (top) is homogeneously distributed throughout the cell (dashed red line). Adapted with permission from ref (10). Copyright 2020 Xiang et al. (g) Confinement is provided by the cell wall in bacteria and by the nuclear envelope in eukaryotes. (i) Possibly due to confinement-induced glassy dynamics, micrometer-sized regions move coherently within the nucleus on a time scale of seconds. Adapted with permission from ref (179). Copyright 2018 Oxford University Press. (ii) Chromosomes territories inside the nuclear confinement. Adapted with permission from ref (180). Copyright 2019 eLife Sciences Publications, Ltd. (iii) Relaxation of the cell-wall confinement (orange line) of E. coli leads to an opening up of the circular bacterial chromosome (white). Adapted with permission from ref (13). Copyright 2019 Springer Nature.
A starting point is to realize that DNA is a very long macromolecule, and hence polymer physics dictates important aspects of its behavior. Bare DNA has a persistence length of 50 nm and can be described by a worm-like chain model.127,128 For genome-sized DNA, this leads to a random polymer coil structure with a sizable radius of gyration, from 3.6 μm for a 4.6 megabasepair circular Escherichia coli bacterial genome to 130 μm for the 3 gigabasepair linear human genome (if it were all to be connected in one linear DNA polymer). Such a random coil is a large and rather open structure of low DNA density that a priori clearly would not fit within the typically available space, which is the set by the ∼1 μm cell size for E. coli(129) (volume of ∼0.5 μm3) and by the ∼10 μm nucleus size for human cells130 (volume of ∼525 μm3). Hence, the DNA needs to be condensed, thereby increasing the DNA density by 2–4 orders of magnitude.
A range of passively acting DNA-binding proteins is available for a first level of condensation (Figure 4a). In eukaryotes, the major binding protein is the nucleosome, which consists of 146 base pairs of DNA wrapped around a histone octamer in 1.7 turns.38 These nucleosomes package DNA into a beads-on-a-string structure, thus compacting DNA by shortening the total polymer length, changing the level of supercoiling,131 and altering flexibility of the DNA fiber.132 Nucleosome-like structures have also been identified in archaea, albeit with different properties as compared to eukaryotes, such as oligomerization.133 In bacteria, DNA-binding proteins known as nucleoid-associated proteins (NAPs) similarly condense the chromosome.39,40 Upon binding the DNA either nonspecifically or at sequence-specific target sites, these NAPs wrap (IHF, Dps), bend (Fis, HU), or bridge (H-NS) the DNA. Aside from the structural role, NAPs also influence gene expression. NAPs such as MatP are implied in the organization and demarcation of the Terminus macrodomain in E. coli, which is flanked by left/right macrodomains that connect to a macrodomain at the origin of replication.134 The mechanism behind the formation of these macrodomains still remains largely unclear. In eukaryotes135 and bacteria,136 post-translational modifications (such as phosphorylation, methylation, and acetylation) of histone tails or NAPs play an important regulatory role by modulating their influence on gene expression and chromosome-structural properties. These modifications work at various levels, as they, for example, change the mutual interactions between DNA-binding proteins which may cause the formation of A/B compartments through phase separation.
Supercoiling43−45 is relevant in both eukaryotes and bacteria (Figure 4b,c). Bacterial genomes exhibit on average negative supercoiling, that is, their DNA is under-twisted compared to the regular right-handed double helix. In all organisms, the local supercoiling is continuously altered by transcribing RNA polymerases that move along the DNA, introducing positive supercoils ahead of them and negative supercoils in their wake.137 In bacteria, transcription processes at highly expressed long genes can lead to both diffusion barriers for supercoils as well as extended decompacted regions that may cause segmentation of the bacterial chromosome into chromosome interaction domains (CIDs).47,138 Control of the supercoiling state happens in two ways: First, passive control of supercoiling is provided by NAPs, such as HU139 and Fis140 which bind at supercoiled plectonemes, thereby stabilizing them. Second, active control of the torsional state of DNA is provided by a variety of topoisomerases141,142 that introduce or relax supercoiling within the DNA. Topoisomerases also play a role in decatenation, thus controlling the topology of the DNA polymer, which is relevant at all stages of the cell cycle, but especially for faithful chromosome segregation in both bacteria and eukaryotes.
A central organizational motif of chromosome structures is DNA looping (Figure 4d). Loops can form if proteins passively bridge two distant points along the DNA.41,42 Alternatively, loops can be produced in an energy-driven process by structural maintenance of chromosomes (SMC) complexes. A wide range of methods (Hi-C,4,101,102 biochemical assays,143−145 and single-molecule experiments146,147) have provided evidence for loop extrusion by SMCs. Direct imaging of the loop extrusion process by a single SMC complex, such as cohesin and condensin, was provided in single-molecule optical visualization assays.5−9 SMCs are motor proteins that bind DNA and then start reeling in the DNA strand, thereby forming a loop. They are fast but weak motors, translocating DNA at rates up to 2000 bp/s but stalling their motor action at forces of less than a piconewton.5−9 The precise molecular mechanism behind SMC loop extrusion is still unknown, although parts of the molecular puzzle are being solved by structural studies with cryo-EM148−151 and dynamical studies with high-speed AFM.152,153 In interphase, cohesin-mediated loops are associated with TADs that often link promoters and enhancers and also correlate with gene activation,3 although the latter is under dispute.154 It is still incompletely understood how the boundaries of TADs are defined in many organisms and how TADs correspond to actual physical structures in single cells.72,73 Metazoan TAD boundaries are often signaled by DNA sites that are bound by CTCF proteins that act as a stop or pause sign for loop extrusion by cohesin.3,155 In preparation of eukaryotic cell division, loop formation by condensin ensures that newly replicated chromosomes are compacted, disentangled, and segregated from each other.54,156 Lastly, in E. coli, Hi-C maps show that the SMC complex MukBEF promotes long-range DNA contacts,49 and live-cell imaging reported that MukBEF occupies a thin axial core within the nucleoid, consistent with a bottle-brush chromosome structure.157
More recently, it has become clear that phase separation likely plays an important role in organizing chromosomes, for example, in the formation of chromosomal compartments and transcriptional hubs, which provide a fast-tunable and selectively accessible environment for gene expression (Figure 4e). Phase separation is often mediated by multiple weak interactions between intrinsically disordered or low-complexity protein domains.158 Attractive interactions between heterochromatin nucleosomes, mediated by histone tails20 or histone-binding proteins35,36 as well as the interaction between heterochromatin and the nuclear boundary or lamina, have been reported to underlie the formation of chromosomal compartments and their organization relative to the nuclear lamina.34,159 The HP1α histone-binding protein, for example, forms liquid droplets in vitro when it is phosphorylated at the N-terminal extension,35 though it did not do so in live mouse cells,160 underlining the need for careful experiments when phase separation is involved.161,162 This process of microphase separation, which segregates the heterochromatin (B-compartmental domains) from the euchromatin (A-compartmental domains), is further modulated by active mixing caused by SMC loop extrusion.24 Zooming in within the A-compartmental domains, transcribed euchromatin may segregate from dormant euchromatin through the formation of active microemulsions with RNA transcripts.26 Chromosomal compartments linked to gene expression levels have also been observed in Sulfolobus archaea, where they correlate with the energy-driven action of an SMC-like protein called coalescin.163 Furthermore, transcriptional hubs in eukaryotes display properties of liquid condensates, where multiple components have been implicated with the phase separation, namely transcription factors,27,28 coactivators,30,31 the enhancer sequence,29 and RNA polymerase.27,28,30,33 Lastly, phase separation is also significant for bacterial chromosomes,164,165 for example, in transcriptional hubs surrounding the nucleoid in E. coli(32) and in ParB protein clusters in B. subtilis.166 ParB loads the bacterial SMCs onto the DNA, whereupon the SMCs actively proceed along the DNA, wrapping the two chromosome arms together.167
Finally, chromosomes are spatially confined within the nucleus (eukaryotes) or cell boundary (bacteria and archaea) and are suspended inside the crowded nucleoplasm or cytosol, respectively (Figure 4f,g). The size and shape of the confinement can strongly impact the chromosome structure. For example, while a spherical container allows mixing of chromosomes, deformation into a cylindrical or disc-like shape may lead to spontaneous demixing and segregation.19 Yet, chromosomes occupy distinct chromosome territories within the roughly spherical nucleus, indicating additional mechanisms. It has been suggested that chromosomes get kinetically trapped into such territories at the start of interphase after the decondensation of mitotic chromosomes.18 Combined with confinement by the cell wall, crowding by macromolecular complexes in the E. coli cytoplasm compacts and positions chromosomes, leading also to a strongly varying ratio between nucleoid size and cell size across bacterial species.13,14,17 Furthermore, the cytoplasm in bacteria is a poor solvent for DNA, causing the spontaneous compaction and formation of domain-like structures.10 Lastly, the DNA polymer itself as well as the surrounding cytoplasm were found to exhibit confinement-induced glassy dynamics, both in bacteria11 and in human cells.12,15,16
Outlook
We reviewed research on chromosome structure and introduced the “genome-in-a-box” (GenBox) as an alternative in vitro approach to build and study chromosomes. GenBox bridges the traditional methodologies of live-cell experiments and in vitro single-molecule studies by using a genome-sized DNA substrate and subsequent addition of DNA-organizing elements. As a method which is based on the principle of ‘to build is to understand’, it will allow to study how local interactions between chromosome building blocks and DNA lead to emergent genome-wide organizing effects. For example, while we know in quite some detail how single SMCs extrude loops of DNA,5−9 it remains unclear how these molecular motors collectively act to form a structured interphase or mitotic chromosome. A GenBox approach enables such studies, while also generally addressing the distinct effects of NAPs, topoisomerases, polymerases, crowding agents, etc. In order to build up further hierarchical levels of complexity, combinations of chromosome building blocks can be probed, since many of these building blocks mutually interact, as detailed in the overview above. In this light, it will be interesting to explore whether it is possible to recreate chromosome-mimicking structures from a minimal set of multiple DNA-structuring elements. For example, a chromosome-mimic together with macromolecular crowders can be placed inside a liposome, which is subsequently shaped with microfabricated structures,104,114 similar to in vivo experiments with shape-manipulated E. coli cells.13,14 Furthermore, we expect a lively interplay with polymer physics modeling, as the GenBox approach is closely related to the typical setup for in silico modeling.
While GenBox will allow a wide array of interesting experiments on genome-sized DNA substrates, no experimental method is without its challenges. For example, this approach does not lend itself well to the discovery of so far unknown building blocks. Hence, like in any in vitro experiment, an attempt to recreate chromosomes with a minimal set of building blocks may fail if a component is missing, indicating the need for a close feedback loop with live-cell experiments. In order to gain access to such a missing component, it may be possible to combine GenBox experiments with cell extracts, that is, combining genome-sized DNA and purified chromosome building blocks with the complexity of the cytoplasm or nucleoplasm of natural cells. Clearly, many technical hurdles will need to be overcome to realize GenBox, for example, to prevent the shearing of the very large and fragile DNA molecules. Quantitation may also pose a challenge as copy numbers of DNA-binding proteins in a cell may not directly translate to in vitro concentrations, since crowding conditions may differ and protein concentrations in cells vary across time as they are under the control of the cell-cycle. In order to study the effects of developmental trajectories and cellular cycles, for example, the transition from interphase to mitotic chromosomes or vice versa, one would need to engineer the ability to temporally control the concentrations of chromosome building blocks. Fortunately, this should be feasible by using microfluidics, in a similar manner to the experiments of Jun et al., who observed compaction and decompaction of isolated chromosomes inside microchannels when crowding agents were added and removed.117
In closing, we like to point out that GenBox is one of multiple avenues that are inspired by research aimed at assembling a synthetic cell. This bottom-up biology approach distinguishes itself from the usual in vitro single-molecule experiments by acknowledging the importance of size, complexity, and collectivity in biological organization and processes. By acting as an intermediary between the current approaches of live-cell experiments and single-molecule techniques, we foresee that GenBox may offer a fruitful avenue to study chromosomes in vitro in a bottom up-manner, yielding valuable insights on chromosome structure and dynamics.
Notes
The authors declare no competing financial interest.
Acknowledgments
We would like to thank Jaco van der Torre, Aleksandre Japaridze, Martin Holub, and Eugene Kim for discussions. We acknowledge funding support from the ERC Advanced Grant LoopingDNA (no. 883684) and the NanoFront and BaSyC programs of NWO-OCW.
Vocabulary
| Bottom up biology | bottom up biology research aims to engineer and study life from the bottom up, from molecules to cells to tissues |
| synthetic cell | synthetic biology research deals with the construction of new biological molecules and systems in order to redesign those found in nature. An ultimate aim in this field is the synthetic cell: to construct an artificial cell-like object that exhibits characteristics of natural cells |
| chromosome organization | the structure of a genome in both the spatial and temporal sense, as it is organized in living systems |
| polymer physics | the physical study of polymers that shows how the global configuration of polymers (e.g., biopolymers such as DNA) is guided by local physical properties such as the stiffness, interactions between different monomers and interactions of the polymer with the surrounding medium |
| chromosome building blocks | DNA-organizing elements, such as DNA-binding proteins or components of the surrounding medium, which interact with and give structure to the genome through a variety of local mechanisms such as bending, bridging, wrapping, looping, crowding, and phase separation. |