
Esophageal adenocarcinoma (EAC) is the dominant form of esophageal malignancies in the United States and other industrialized countries. The incidence of EAC has been rising rapidly during the past four decades. Barrett's esophagus (BE) is the main precancerous condition for EAC, where a metaplastic columnar epithelium replaces normal squamous mucosa of the lower esophagus. The primary risk factor for BE and EAC are chronic gastroesophageal reflux disease (GERD), obesity and smoking. During the BE-dysplasia-EAC sequence, esophageal cells are under a tremendous burden of accumulating reactive oxygen species (ROS) and oxidative stress. While normal cells have intact antioxidant machinery to maintain a balanced anti-tumorigenic physiological response, the antioxidant capacity is compromised in neoplastic cells with a pro-tumorigenic development antioxidant response. The accumulation of ROS, during the neoplastic progression of the GERD-BE-EAC sequence, induces DNA damage, lipid peroxidation and protein oxidation. Neoplastic cells adapt to oxidative stress by developing a pro-tumorigenic antioxidant response that keeps oxidative damage below lethal levels while promoting tumorigenesis, progression, and resistance to therapy. In this review, we will summarize the recent findings on oxidative stress in tumorigenesis in the context of the GERD-BE-EAC process. We will discuss how EAC cells adapt to increased ROS. We will review APE1 and NRF2 signaling mechanisms in the context of EAC. Finally, we will discuss the potential clinical significance of applying antioxidants or NRF2 activators as chemoprevention and NRF2 inhibitors in treating EAC patients.
| ABS | acidic bile salts |
| ARE | antioxidant response elements |
| ALDH1 | aldehyde dehydrogenase 1 |
| APE1 | Apurinic/apyrimidinic endonuclease 1 |
| AP-1 | activator protein-1 |
| ATBC | Alpha-Tocopherol Beta Carotene |
| BE | Barrett's esophagus |
| BER | base excision repair |
| CAT | catalase |
| CSC | cancer stem cell |
| COSMIC | Catalogue of Somatic Mutations in Cancers |
| CREB | cAMP response element binding protein |
| DMF | dimethyl fumarate |
| ER | endoplasmic reticulum |
| EAC | esophageal adenocarcinoma |
| GERD | gastroesophageal reflux disease |
| GSK3 | glycogen synthase kinase-3 |
| GPx | glutathione peroxidase |
| GR | glutathione reductase |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| GSTM | glutathione S-transferase mu |
| GSTT | glutathione S-transferase theta |
| GSTP | glutathione S-transferase pi |
| HO-1 | heme oxygenase-1 |
| H2O2 | hydrogen peroxide |
| 4-4-HNE | hydroxy-2-nonenal |
| HIF-1α | hypoxia-inducible factor-1α |
| JNK | c-Jun N-terminal kinases |
| KEAP1 | kelch‐like‐ECH‐ associated protein 1 |
| MDA | malondialdehyde |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor κB |
| NRF2 | nuclear factor erythroid 2‐related factor 2 |
| NFE2L2 | nuclear factor erythroid 2 like 2 |
| NQO1 | NAD(P)H quinone oxidoreductase 1 |
| 8-OHdG | 8-hydroxydeoxy-guanosine |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| STAT3 | signal transducer and activator of transcription 3 |
| SOD | superoxide dismutase |
| CuZn-SOD | copper–zinc SOD |
| Mn-SOD | manganese SOD |
| EC-SOD | extracellular-SOD |
| SELECT | Selenium and Vitamin E Cancer Prevention Trial |
| UGT | UDP-glucuronosyltransferases |
| USPSTF | U.S. Preventative Services Task Force |
Oxidative stress results from an imbalance between the production of oxidants that are free radicals and reactive metabolites, including reactive oxygen species (ROS), reactive nitrogen species (RNS), and anti-oxidants that can eliminate ROS or RNS [1]. ROS are products of normal cellular metabolism processes. Mitochondria, where the electron transfer to molecular oxygen occurs at the respiratory chain level, is the site to generate major ROS [2,3], such as superoxide anion (O2 −), hydrogen peroxide (H2O2), hydroxyl radical (OH•-) as well as organic peroxides as normal product of the biological reduction of molecular oxygen. In addition, enzymatic reactions catalyzed by oxidases such as NADPH oxidases, cyclooxygenases, lipooxygenases, and peroxisomal oxidative metabolism, endoplasmic reticulum (ER) stress all generate ROS [2,3]. Under normal physiological conditions, cells have intact anti-oxidant systems to keep ROS tightly controlled at a low level (Fig. 1A). ROS is involved in multiple signaling pathways, such as proliferation and differentiation. ROS plays a vital role in the cellular response to various intracellular environmental stimuli and extra-cellular environmental stimuli [4]. However, accumulation of ROS/RNS causes oxidative stress that poses potential damage to the cells by attacking macromolecules such as DNA, RNA, proteins and lipids. For example, oxidative stress leads to oxidative DNA damage, forming 8-hydroxydeoxy-guanosine (8-OHdG), the hydrolysis product of 8-hydroxyguanosine [5]. The 8-OHdG is a pro-mutagenic lesion that mispairs with adenine, leading to GC to TA transversion [6]. The 8-OHdG is the most widely used oxidative DNA/RNA damage marker [7,8] that has been strongly implicated in carcinogenesis [[9], [10], [11]].

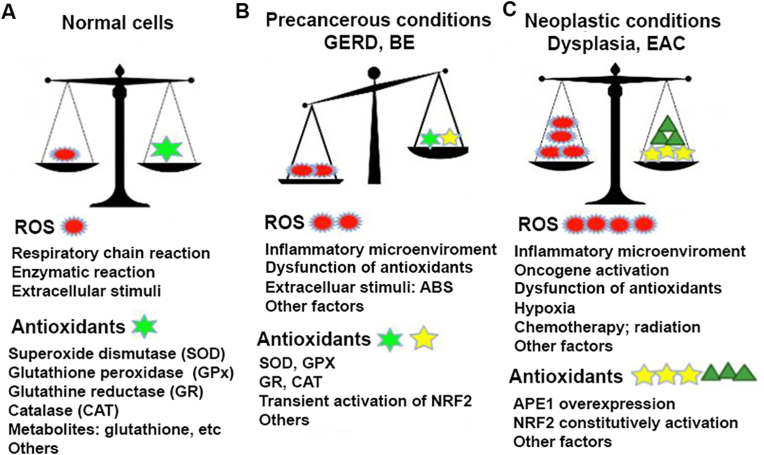
Redox balance in normal, precancerous and neoplastic esophagus.
A) ROS are by-products of normal cellular metabolism and activities. The mitochondria is the major site to produce ROS through the respiratory chain reactions (oxidative phosphorylation). Some enzymatic reactions such as NADPH oxidases, cyclooxygenases, lipooxygenases as well as peroxisomal oxidative metabolism, endoplasmic reticulum (ER) stress all generate ROS. However, normal cells have intact antioxidant machinery including antioxidant enzyme systems such as SOD, GPX, CAT, GR and antioxidant metabolites to keep ROS/antioxidants balance in low level. B) Under GERD, Barrett's esophagus (BE) conditions, refluxes of acidic bile salts, inflammatory microenvironment and activation of inflammation promotion signaling pathways like NF-kB, Stat3 result in elevated ROS generation and accumulation. On the other hand, some of antioxidant enzymes, such as GPX3, GPX7 lost partial function. Consequently, oxidative stress often occur that leads to DNA damage and other cellular damage. C), In neoplastic esophagus (high grade dysplasia and EAC), ROS level further elevate due to mainly the activation of oncogenes, growth factors and fully dysfunction of some antioxidant enzymes (GPX3, GPX7). To avoid cell death due to high ROS level, neoplastic cells develop a pro-tumorigenic antioxidant mechanism by constitutively activating APE1-NRF2 master antioxidant machinery. The hijack of NRF2 by tumor cells regain redox balance in higher ROS levels that favors tumor cell survival.
Esophageal Cancer is the 7th most frequent human malignancies and remains the 6th cancer-related death worldwide [12]. There are two major histology types of Esophageal Cancer: Squamous Cell Carcinoma (SCC) and adenocarcinoma [13]. While the incidence of squamous cell carcinoma is declining in the Western world during the past few decades, the prevalence of esophageal adenocarcinoma (EAC) has been rising rapidly in the US and other Western countries, becoming the dominant form of Esophageal Cancer [[14], [15], [16]]. EAC has three major risk factors: gastroesophageal reflux disease (GERD), obesity and smoking [16]. GERD is a condition in which the gastric contents mixed with bile salts reflux into the lower esophagus, causing acute and chronic inflammatory diseases [17]. Chronic GERD leads to Barrett's Esophagus (BE) development, where the original esophageal squamous epithelium is replaced by a specialized metaplastic columnar epithelium [18,19]. BE is the major known precancerous condition with a risk of progression to low-grade dysplasia, high-grade dysplasia and finally, EAC [20,21]. Accumulating lines of evidence indicate that oxidative stress is involved in the GERD-BE-EAC progression cascades [[22], [23], [24]]. We will review oxidative stress in the context of Barrett's related tumorigenesis. We will discuss the regulatory mechanisms and potential clinical significance of antioxidant activators as chemoprevention approach and inhibitors in EAC treatment.
Recurrent episodes of reflux of gastric juice and bile salts mixture into the lower esophagus are the unique characteristics of GERD-BE and EAC spectrum. We have demonstrated that a short-time exposure of esophageal cells to the acidic bile salts (ABS) to mimic a GERD episode led to significant generation of H2O2 levels and rapid elevation of intracellular ROS levels [22,[25], [26], [27], [28]]. Subsequently, oxidative DNA damage levels (detected by 8-OHdG) and double strand DNA breaks (detected by γH2AX) were significantly increased after ABS exposure [22,25]. EAC is considered a model of oxidative stress and inflammation-driven cancer [29,30]. Chronic GERD induces chronic inflammation (esophagitis) that persists through the BE and EAC progression cascade. During the inflammation process, various inflammatory cells, including mast cells, macrophages and leukocytes, are recruited to the site. As a result, an increased uptake of oxygen, so-called a “respiratory burst,” amplifies the release and accumulation of ROS at the site of damage [1,31].
The inflammatory cells produce various cytokines, chemokines and metabolites as soluble mediators, which promote further recruitment of inflammatory cells to the site of damage with more ROS production. We and others have demonstrated that ABS exposure and inflammatory cytokines and chemokines activate signal transduction cascades such as nuclear factor κB (NF-κB), signal transducer and activator of transcription 3 (STAT3), and hypoxia-inducible factor-1α (HIF-1α) [30,[32], [33], [34]]. Activation of these oncogenic networks contribute to the generation of additional ROS. In line with the experimental observations, the significant increase in ROS levels and oxidative DNA damage in BE-related lesions is confirmed in several experimental animal models [23,35] as well as in human BE and esophageal adenocarcinoma tissue samples [[36], [37], [38]] (Fig. 1).
Normal cells possess intact anti-oxidant machinery to keep ROS level tightly controlled to counteract oxidative stress (Fig. 1A). The balance of oxidants/anti-oxidants is pivotal to maintain normal cellular functions and homeostasis [[39], [40], [41]]. The cellular anti-oxidant defense is comprised of the major enzymatic anti-oxidants that are directly involved in the neutralization of ROS (Fig. 1). Examples include superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GR), and catalase (CAT) [42,43]. SOD is the first characterized anti-oxidant enzyme that includes three types: copper–zinc SOD (CuZn-SOD), manganese SOD (Mn-SOD), and extracellular-SOD (EC-SOD). All of three can catalyze the dismutation of superoxide anion radical (O2 •-) into hydrogen peroxide (H2O2) by reduction [44]. The produced H2O2 is broken into water and oxygen (O2) by catalase (CAT) or glutathione peroxidase (GPx). The GPx enzymes remove H2O2 through oxidizing reduced glutathione (GSH) into oxidized glutathione (GSSG). Glutathione reductase (GR) regenerates GSH from GSSG, using NADPH [42,43].
In addition, some metabolites, such as l-arginine, glutathione, coenzyme Q10, uric acid, lipoid acid, bilirubin, metal-chelating proteins, transferrin, and melatonin also play an essential role in cellular anti-oxidant defense [41,45].
Several genetic variations have been linked to the susceptibility to Barrett's esophagus [[46], [47], [48]]. Among genes encoding anti-oxidant enzymes, variants of GSTP1 (such as rs1695 A>G missense variant), results in reduced enzymatic activity, frequently linked to risks of BE and EAC [49,50]. Variants of GSTT2 were different in African Americans, as compared with European Americans. These different variants are associated with higher expression of the enzyme in African American. The findings may explain the inherent different susceptibility risk to Barrett's esophagus in the population [51]. Furthermore, there are accumulating lines of evidence showing that the cellular anti-oxidants capacity is compromised during BE-EAC tumorigenesis (Fig. 1B) [[52], [53], [54], [55]]. The first line of anti-oxidant enzyme, Mn-SOD, is downregulated in BE and EAC [55]. Several studies have demonstrated a significant reduction in the levels of glutathione contents with major glutathione S-transferases. We have shown frequent DNA hypermethylation and downregulation of multiple anti-oxidant enzymes in BE and EAC. These include glutathione peroxidases (GPX3 and GPX7), glutathione S-transferases (such as GSTM2, GSTM3, GSTM5) [56,57] and metallothionein 3 (MT3) [58]. Dysfunction of these anti-oxidant enzymes makes esophageal cells more sensitive to ABS exposure. It promotes oxidative stress and subsequent DNA damage [25]. Notably, some of these anti-oxidant enzymes such as GPX3 and GPX7 possess unique tumor suppressor functions, in addition to their anti-oxidant properties. For example, GPX3 has a tumor suppressor role in esophageal adenocarcinoma [59], gastric cancer [60,61], breast cancer [62], prostate cancer [63], and colorectal cancer [64]. Similarly, GPX7 has anti-tumorigenic functions in esophageal [57], and gastric adenocarcinomas [65]. Therefore, dysfunction of these antioxidant enzymes not only compromises cellular antioxidant capacity but also favors tumor cell growth, a major contribution to Barrett's tumorigenesis [57,66].
It is well documented that cancer cells possess higher ROS levels than normal or pre-cancerous cells [1,37,67,68]. Factors contributing to increasing ROS in cancer cells include activation of oncogenes (such as RAS and MYC), inflammatory microenvironments, and dysfunction of normal anti-oxidant enzymes. Other factors include extracellular stimuli such as exposure to reflux conditions, infection with H. pylori, radiation, and drug treatments (Fig. 1 C). However, excessive accumulation of ROS and subsequent oxidative DNA damage will incur serious DNA damage response that is lethal to cancer cells. Cancer cells must evolve to develop pro-tumorigenic anti-oxidant mechanisms that maintain oncogenic activities while keeping ROS and oxidative stress below lethal levels.
Nuclear factor erythroid 2‐related factor 2 (NRF2) is a master antioxidant transcription factor playing a central role in regulating the expression of hundreds of antioxidant genes to defend against toxic and oxidative insults [[69], [70], [71]]. NRF2 is encoded by NFE2L2 (Nuclear Factor, Erythroid 2 Like 2) gene and belongs to the Cap'n’Collar (CNC) subfamily of basic leucine zipper (bZIP) transcription factors. NRF2 is constantly undergoing rapid ubiquitination and proteasomal degradation through its physiological inhibitor, KEAP1 (Kelch‐like‐ECH‐ associated protein 1) [72,73] (Fig. 2A). However, when cells are under oxidative stress, the cysteine residues in KEAP1 are oxidized, leading to KEAP1 conformational changes that result in the release of NRF2 from KEAP1. As a result, the free and newly synthesized NRF2 is protected from KEAP1-mediated ubiquitination and degradation. It accumulates and translocates to the nucleus where it binds to the antioxidant response elements (ARE) on the promoter region of its target genes (Fig. 2B). More than 250 NRF2 target genes were reported, including many genes that directly or indirectly possess antioxidant properties. Examples of NRF2 target genes include aldehyde dehydrogenase 1 (ALDH1), NAD(P)H quinone oxidoreductase 1 (NQO1), glutathione S-transferases (GSTs), UDP-glucuronosyltransferases (UGTs), heme oxygenase-1 (HO-1), and glutathione reductase (GR) [74,75]. In addition to the canonical KEAP1-dependent activation of NRF2, several KEAP1-independent mechanisms regulate NRF2 expression and stability [74,76] (Fig. 2B). GSK3- α/β has been recognized as another NRF2 negative regulator by targeting NRF2 to the SCF/β-TrCP-mediated degradation, which is reported to be more sensitive to metabolic changes [[77], [78], [79]]. HRD1, regulated by XBP1 [80], an endoplasmic reticulum protein, has recently been reported as a novel E3 ubiquitin ligase responsible for compromised NRF2 response during liver cirrhosis [81]. A number of proteins can bind to KEAP1, disrupt the KEAP1-NRF2 complex, leading to accumulation of NRF2, such as p62, an autophagy-related protein [82,83], dipeptidyl peptidase 3 (DPP3) [84], Wilms tumor gene on X chromosome (WTX) [85], and Partner and localizer of BRCA2 (PALB2) [86]. In addition, p21Cip1/WAF1 [87] and BRCA1 [88] can directly interact with NRF2, thereby disrupting the KEAP1-NRF2 complex to activate NRF2.

NRF2 regulatory mechanisms.
A) three reported negative regulatory mechanisms of NRF2. KEAP1 is the first and major NRF2 negative regulator in cells. In normal condition, KEAP1 binds to NRF2 and connects NRF2 to Cullin3 E3 ubiquitin ligase, leads to NRF2 degradation in proteasome. In addition, GSK-3α/β kinases can phosphorylate NRF2 which lets it binding to β-TrCP-Cul1 ubiquitin ligase, leads to NRF2 degradation in proteasome. HRD1 is a recently identified E3 ubiquitin ligase which targets NRF2 when activated by XBP1 under ER stress. So far, this regulatory pathway has only been reported during liver cirrhosis. B) NRF2 activation mechanisms. When cells are under stresses such as electrophilic stimuli, cysteine molecules in KEAP1 are modified (oxidized) that leads to release of NRF2 from KEAP1-Cul3-mediated ubiquitination. The free and newly-synthesized NRF2 then accumulates and translocates to nucleus and binds to the ARE on its target genes' promotor and activates its target genes. So far, about 250 target genes have been reported involving Phase I-III metabolism, redox regulation, lipid and carbohydrate metabolism, iron/hemo metabolism and many other cellular processes. In addition to this canonic KEAP1 dependent mechanism, many non-canonic mechanisms have been reported. For example, KEAP1 genetic mutations, promoter hypermethylation which inactivate KEAP1, some proteins binding to KEAP1 to interfere with KEAP-NRF2 interaction, such as p62, PALB2. Genetic mutations of NRF2, epigenetic regulation, phosphorylation and SUMOylation of NRF2 itself activate NRF2. Some proteins such as p21, BRCA1 and APE1 have been reported to bind to NRF2 to interfere with NRF2-KEAP1 interaction. Taken together, the NRF2 is a master transcriptional regulator in cellular antioxidant function and other cellular processes. The regulatory mechanisms are complex and context-dependent.
Although NRF2 activation may reduce cancer risk by suppressing oxidative stress and tumor promoting inflammation, an increase in NRF2 expression and activity level has been observed in several cancer types [71,72,75]. The constitutive activation of NRF2 has been reported in several cancers through NFE2L2 genetic mutations [89,90]. We have shown that transient exposure of EAC cells to reflux conditions induced NRF2 accumulation and activation with upregulation of its target genes [91]. The induction of HO-1 and GR in these cells protect against ABS-induced oxidative DNA damage and apoptosis, in concordance with NRF2 basic antioxidant functions [91]. We observed that NRF2 was constitutively upregulated in neoplastic esophageal cells (dysplasia and EAC) and primary EAC samples. Surprisingly, NRF2 was not constitutively up-regulated in non-neoplastic Barrett's cells and tissues. Our analysis of COSMIC (Catalogue of Somatic Mutations in Cancers) database indicated low incidence of mutation of NFE2L2 and KEAP1 in EAC (NFE2L2 in 6.6% (38/576) whereas KEAP1 in 1.22% (7/576)). Therefore, the frequent constitutive overexpression of NRF2 in EAC is a non-mutational event that is likely independent of canonical KEAP1 mechanisms.
The Apurinic/apyrimidinic endonuclease 1 (APE1) (also known as APEX1 or redox factor 1 (REF1)) is one of the key enzymes of the base excision repair (BER) pathways in mammals. APE1 is the key enzyme required for repair of Apurinic/apyrimidinic (AP) sites, a major type of oxidative DNA damage lesions generated by ROS [[92], [93], [94]]. On the other hand, APE1 possesses redox activity that is required for activation of redox-dependent transcription factors such as HIF-1α (hypoxia inducible factor-1α) [95], NF-κB (nuclear factor-κB (NF-κB) [96], p53, CREB (cAMP response element binding protein) and AP-1 (activator protein-1) [94,97]. Recent studies indicate that APE1 is frequently overexpressed at the protein level in human cancers, including EAC [[98], [99], [100]]. APE1 was also induced in response to ABS exposure in esophageal cells [100]. High levels of APE1 protect Barrett's and EAC cells against oxidative DNA damage and DNA double stand breaks, promoting cell survival by regulating JNK and p38 kinases [100]. We have demonstrated that the APE1 redox activity is required for ABS-induced activation of EGFR-STAT3 signaling axis [33]. Activation of STAT3 is an important molecular landmark in carcinogenesis for many human malignancies [101,102], including gastrointestinal tract cancers [103]. Furthermore, APE1 interacts with ARF6 to enhance MMP14 recycling in a redox-sensitive manner, promoting cell invasion [104]. Of note, our recent work suggests that APE1 may regulate NRF2 protein expression in esophageal epithelial cells through a KEAP1-independent regulation of NRF2 stability, likely dependent on APE1 redox activity in EAC (unpublished data). Based on these multiple lines of evidence, the constitutive upregulation of APE1-NRF2 axis in Barrett's carcinogenesis rebalances redox homeostasis in favor of tumorigenic cell survival, contributing to tumor progression and invasion (Fig. 1 C).
As we have mentioned above, during GERD-BE-EAC process, episodes of reflux of acidic bile salts and chronic inflammation induce high levels of ROS [24,105]. Oxidative stress leads to oxidative DNA damage in both nuclear and mitochondrial DNA, including DNA strand breaks [25,27,28]. In addition, ROS induces protein oxidation as well as lipid peroxidation, producing mutagenic and carcinogenic reactive aldehydes such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) [[106], [107], [108]], known to be associated with tumorigenesis [109]. We have demonstrated that upon ABS exposure, esophageal cells respond by transiently activating APE1 and NRF2 pathways to reduce intracellular ROS and subsequent DNA damage [91,100]. Therefore, NRF2 may play a protective tumor suppressor role during the early pre-neoplastic stages of esophageal tumorigenesis (Fig. 3A). This is consistent with early discoveries using NRF2 knockout mice which demonstrated that NRF2 knockout mice were more susceptible to chemical-induced tumorigenesis [110,111].
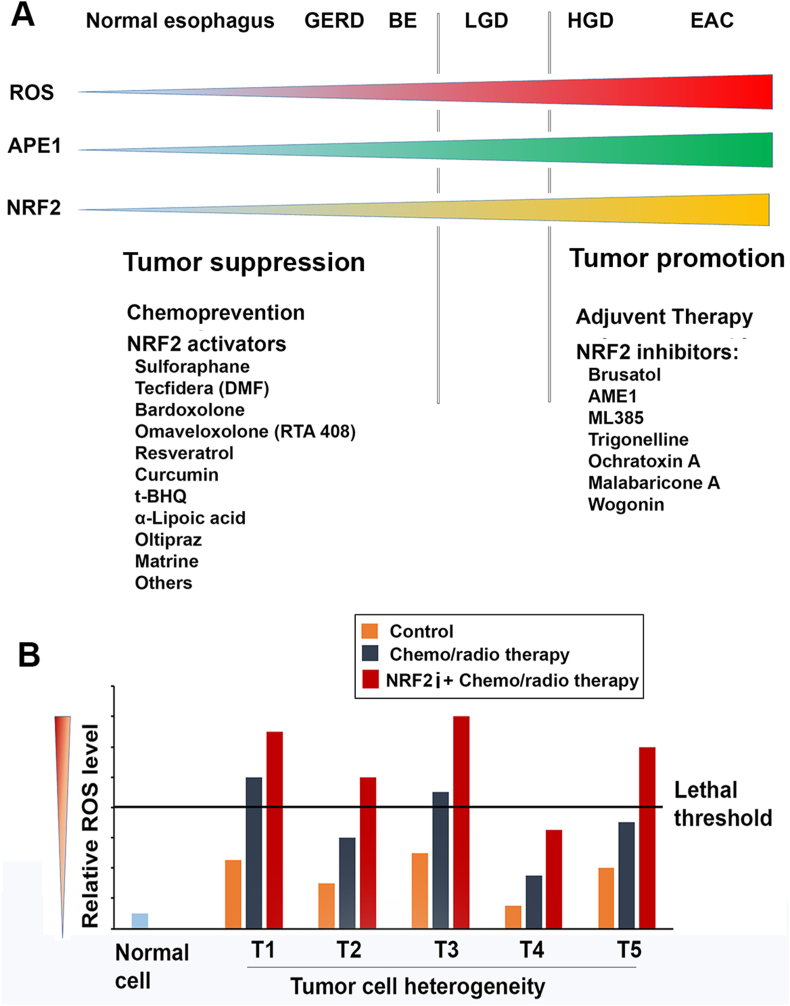
Potential application of NRF2 activators and inhibitors in GERD-BE-EAC patients.
A) Based on ROS, APE1 and NRF2 levels in GERD-BE-EAC sequence, it is expected that NRF2 activators may be benefit for the patients with GERD and BE. However, cautions must be taken due to the “Dark Side” of NRF2 in which activated NRF2 may promote tumor growth once tumor initiating cells form. For EAC patients, NRF2 inhibitors may be used as adjuvant therapy or in combination with other therapeutic approaches. B) An assumptive figure to illustrate potential mechanisms of use NRF2 inhibitors (NRF2i) in combination with other therapeutic approaches, particularly those that work on the increase of ROS to kill cancer cells, for example cisplatin. Primary cancer cells usually are heterogeneity in terms of intracellular ROS level. Some cancer cells such as cancer stem cells or senescent cells may possess lower ROS than regular cancer cells. Therefore, single drug therapy may only kill partial cancer cells with higher basal ROS levels. Administration of NRF2 inhibitors (NRF2i) in combination with other chemo-/radio-therapy sensitizes tumor cells to these therapies through obvious increase ROS level to lethal threshold. T1-T5 represent tumor populations with different ROS levels.
However, in neoplastic esophageal cells (dysplastic and EAC), NRF2 is constitutively overexpressed. High levels of NRF2 in neoplastic cells counteract elevated ROS levels and promotes tumor cell survival [100,105]. The overexpression of NRF2 has been associated with uncontrolled tumor growth, angiogenesis, migration, invasion, metastasis and drug resistance [69,75,112]. Recent discoveries indicate that NRF2 is an important player in reprograming cancer cell metabolism [[113], [114], [115]], cancer stem cell properties [116,117], tumor microenvironment and immune response [[118], [119], [120]]. ALDH1 (Aldehyde dehydrogenase 1), an enzyme normally involved in retinol metabolism catalyzing/oxidizing retinaldehyde to retinoic acid, is found upregulated in many human cancer types including gastrointestinal cancers [121,122] and is associated with poor prognosis [123,124]. More importantly, ALDH1 is regarded as one of the unique cancer stem cell (CSC) markers of many human malignancies, and is required for tumor cells to maintain their stemness [124,125]. Heme oxygenase-1 (HO-1) is a microsomal enzyme which catalyzes the oxidation of heme to biologically active products: carbon monoxide (CO), biliverdin, and ferrous iron [126]. As a NRF2 target gene, we have shown that HO-1 was transiently induced in esophageal epithelial cells in response to ABS exposure conditions that mimic a GERD episode [91]. However, like NRF2, HO-1 is overexpressed in many human cancers [127,128]. Constitutive upregulation of HO-1 in tumor tissues favors tumor cells' survival through various mechanisms that include decreasing intracellular ROS, activating AKT and inhibiting apoptosis pathways [128,129]. NRF2 plays a key role in activation of almost all known hallmarks of cancer [129]. In almost all cancer types, overexpression NRF2 is associated with poor prognosis and drug- or radio-resistance [130,131]. In this context, NRF2 plays an oncogenic role in EAC once tumor initiating cells are formed (Fig. 3A). Based on current lines of evidence, we can speculate that silencing of several genes in the glutathione pathways during Barrett's tumorigenesis is compensated via overexpression of NRF2 and possibly APE1 to promote a tumorigenic anti-oxidant response. This could be especially important with the recent evidence showing that some of the silenced glutathione family members, such as GPX3 and GPX7, possess tumor suppressor functions (discussed above). The molecular switch from transient induction to constitutive overexpression of NRF2 may define a conversion from an anti-tumorigenic to pro-tumorigenic antioxidant response during Barrett's carcinogenesis.
Oxidative stress, associated with tumorigenesis, is linked with aging and many human diseases such as inflammatory diseases, neurodegenerative disorders and other diseases [1,[132], [133], [134]]. Therefore, a great effort has been made to explore various antioxidants for prevention or treatment of oxidative stress-related diseases [40,45,135]. In terms of antioxidants in cancer prevention and treatment, numerous per-clinical investigations and clinical trials have been conducted (https://www.clinicaltrials.gov/). It was expected that anti-oxidants would reduce risks for development of cancers. On the contrary, some trials showed that anti-oxidants increased the risk of developing some cancers such as lung cancer [136] and prostate cancer [137,138]. For example, the Alpha-Tocopherol Beta Carotene (ATBC) Cancer Prevention Study reported that male smokers receiving b-carotene had an 18% increase in lung cancer incidence [136]. The SELECT trial [137,138] was a randomized, double-blind, placebo-controlled trial of older males given vitamin E, selenium, both, or neither for 7–12 years. This large trial reported that men taking vitamin E alone had significantly higher risk to develop prostate cancer. Gill et al. [67] recently summarized randomized, placebo-controlled trials that included more than 10,000 participants. The U.S. Preventative Services Task Force (USPSTF), reviewing many of the available clinical trials, concluded that there was insufficient data for or against the use of most nutrient supplements for cancer prevention [139]. Similarly, clinical trials using anti-oxidants as adjuvant therapy in cancer patients’ treatment showed either no clear benefit or contradictory results [140,141].
As NRF2 is recognized as the master regulator of cellular anti-oxidative properties, numerous efforts have been made in developing NRF2 activators or inhibitors [142]. Hundreds of investigations have conducted in vitro and in vivo to evaluate natural or synthesized compounds as NRF2 activators or inhibitors [69,74,143]. So far, only fumaric acid ester dimethyl fumarate (DMF) (also called BG-12 or Tecfidera, from Biogen) has been FDA approved for treatment of psoriasis for many years [144]. Tecfidera was also approved in 2013 to treat relapsing-remitting multiple sclerosis (MS) [[145], [146], [147]]. Oltipraz (4-methyl-5(pyrazinyl-2)-1-2-dithiole-3-thione) is another promising NRF2 inducer undergoing evaluation in a phase III clinical trial for the treatment of nonalcoholic fatty liver disease [148]. In addition, several natural compounds such as sulforaphane [149,150], curcumin [151,152], and resveratrol [153,154] have been identified as NRF2 activators and are under extensive clinical trials in a wide range of oxidative-stress diseases (https://www.clinicaltrials.gov/). However, in terms of using these NRF2 inducers in cancers, a few have been tested in clinical trial studies. The major concern for modulating NRF2 using NRF2 activators relies on the fact that NRF2 functions are hijacked by tumor cells to favor their growth and progression [70,71,155,156]. This so-called “Dark Side” of NRF2 makes it a double-edged sword: a must-caution before applying these compounds in patients. Once tumor-initiating cells are formed at early stage of tumorigenesis, the administration of these compounds may accelerate tumor progression, even promoting metastasis [157,158] (Fig. 3A). For example, Tao, et al. [159] tested modulation of NRF2 in several in vivo mouse models and demonstrated that NRF2 activation prevented initiation of chemically induced cancer, and promoted progression of pre-existing tumors regardless of a chemical or genetic etiology. On the other hand, because NRF2 is overexpressed in many human cancers and is associated with tumor progression, metastasis, drug- or radio-resistance, and poor survival, targeting NRF2 has become an attractive adventure in cancer therapy. Brusatol (quassinoid, natural) is the first NRF2 inhibitor identified by screening natural product extracts [160]. Brusatol is reported to sensitize cancer cells to chemotherapeutic drugs via inhibition of NRF2 signaling pathway [160,161] (Fig. 3B). Although it has been pointed out that brusatol is a global translation inhibitor, it remains a useful tool to study NRF2 inhibition in cancer. Proteins with short half-life, such as NRF2, are the most susceptible ones to inhibitors of protein translation [162]. Tao et al. [159] reported that once tumors are initiated, NRF2 inhibition by brusatol is effective against chemically and spontaneously induced tumors. In addition to brusatol, other investigational compounds are capable to inhibit NRF2 activity, such as AME1 [163], ML385 [164], trigonelline [165], malabaricone A [166], ochratoxin A [167], and wogonin [168]. However, the molecular mechanisms and biological effects of these compounds need to be further investigated.
In terms of GERD-BE and EAC cascade (Fig. 3), as discussed above, we and others have demonstrated increased oxidative stress in this process. We reported that NRF2 is pivotal for esophageal epithelial cells to battle oxidative stress and protect cells from ABS-induced DNA damage. Therefore, from a hypothetical point of view, NRF2 activators may benefit patients and reduce risks for EAC development. However, a word of caution is due given the pro-tumorigenic functions of NRF2. NRF2 activators may in fact accelerate the progression of pre-neoplastic and neoplastic cells towards EAC. It is worth mentioning that there are reports of a beneficial role of antioxidants, such as manganese superoxide dismutase [169]. Several investigations reported that dietary antioxidants, fruits, and vegetables are inversely associated with BE or EAC risk [170,171], while no association was observed for supplements intake [170]. Of note, an early system review and meta-analysis of 14 randomized trials failed to find significant effects of supplementation with b-carotene, vitamins A, C, E, and selenium (alone or in combination), versus placebo, on esophageal, gastric, colorectal, pancreatic, and liver cancer incidences [172]. Regarding NRF2 activators as chemoprevention for BE and EAC, only few reports are available [173,174]. Some studies suggest that resveratrol [173] or curcumin supplementation [174] may have a chemoprevention role in BE. These findings exemplify the dilemma surrounding antioxidants and NRF2 applications in cancer prevention and treatment. Considering the so-called “Dark Side of NRF2”, there are warranted serious concerns for administering antioxidants or NRF2 activators in Barrett's conditions, where the cellular context may significantly alter the response from an anti-tumorigenic to a pro-tumorigenic outcome. For NRF2 inhibitors in EAC, these have not been fully investigated and there are no FDA approved drugs. In fact, the clinical utility of the pre-clinical studies is limited. These studies provide a proof of concept, not ready for clinical trials due to several limiting properties of the existing inhibitors.
The antioxidant response in Barrett's tumorigenesis is a complex molecular process. The observed molecular switch from transient induction of some antioxidant genes in pre-neoplastic conditions to their constitutive overexpression in neoplastic cells highlight different molecular functions and biological outcomes. Antioxidants such as APE1 and NRF2 may function as a double-edged sword where the outcome is context dependent where the stimulus and the cellular state matter. The molecular mechanisms and target genes that alter the anti-versus pro-tumorigenic functions of antioxidant response need to be fully elucidated to determine if and when activators or inhibitors can be utilized in patients with Barrett's or EAC.
None.
1
2
3
4
6
7
8
9
10
11
12
13
14
15
16
20
21
22
23
24
25
26
27
28
29
32
33
34
35
36
37
38
39
40
41
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
71
72
73
74
76
77
78
79
80
81
83
84
85
86
87
88
89
90
91
92
93
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
157
158
159
160
161
163
164
165
166
167
168
169
170
171
172
173
The research reported in this publication was supported by grants from the
 The antioxidant response in Barrett's tumorigenesis: A double-edged sword
The antioxidant response in Barrett's tumorigenesis: A double-edged sword
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
