
DJ-1 is a multifaceted protein with pleiotropic functions that has been implicated in multiple diseases, ranging from neurodegeneration to cancer and ischemia-reperfusion injury. Ischemia is a complex pathological state arising when tissues and organs do not receive adequate levels of oxygen and nutrients. When the blood flow is restored, significant damage occurs over and above that of ischemia alone and is termed ischemia-reperfusion injury. Despite great efforts in the scientific community to ameliorate this pathology, its complex nature has rendered it challenging to obtain satisfactory treatments that translate to the clinic. In this review, we will describe the recent findings on the participation of the protein DJ-1 in the pathophysiology of ischemia-reperfusion injury, firstly introducing the features and functions of DJ-1 and, successively highlighting the therapeutic potential of the protein.

•
DJ-1 has been shown to confer protection in ischemia-reperfusion injury models.
•DJ-1 protection relies on the activation of antioxidant signaling pathways.
•DJ-1 regulates mitochondrial homeostasis during ischemia and reperfusion.
•DJ-1 seems to modulate ion homeostasis during ischemia and reperfusion.
•DJ-1 may represent a promising therapeutic target for ischemia-reperfusion injury.
DJ-1 is a 189 amino-acid protein and exists as a homodimer of 20 kDa highly conserved across phyla [1]. Alterations in the protein functionality have been associated with different human diseases, ranging from Parkinson's disease, cancer, male infertility, diabetes, stroke, chronic obstructive pulmonary disease, and ischemia-reperfusion (IR) injury [2,3]. The role of DJ-1 in several diseases reflects its wide pattern of expression, being found in most body tissues [4]. At the subcellular level, DJ-1 is localized primarily in the cytoplasm, though it has also been found to translocate to the nucleus and mitochondria, in particular, under conditions of stress [[5], [6], [7], [8]]. In addition, DJ-1 has been shown to be secreted into the extracellular space in several pathologies, as it has been detected in the plasma or serum of patients affected by melanoma, breast cancer, and stroke [[9], [10], [11]].
Despite the great efforts in the scientific community, the physiological function of DJ-1 is only partially understood. To date, several roles have been ascribed to the protein, including tumorigenesis, modulation of signaling cascades, preservation of ROS homeostasis, regulation of transcription, maintenance of glucose levels, and protection against protein aggregation [12,13]. Nonetheless, the most widely accepted function of DJ-1 is its protective role against excessive ROS levels [12,13]. In this regard, it has been suggested that DJ-1 may sense the cellular redox state through a highly conserved cysteine (Cys) residue, localized at position 106 [14]. Indeed, due to its low thiol pKa value, this residue exists almost exclusively as a reactive thiolate anion at physiological pH and it has been reported to be particularly sensitive to oxidation, probably regulating several functions attributed to the protein [15]. In the antioxidant defense, DJ-1 has been reported to orchestrate a large variety of responses to promote cell survival. In this frame, it has been shown that DJ-1 can indirectly affect transcription by interacting with transcription factors and their regulators, thus modulating gene expression [16]. In particular, the protein often leads to the activation of pro-survival and proliferative pathways, such as the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) [17], the nuclear factor erythroid 2-related factor 2 (Nrf2) [18], and the Akt pathways [19,20], while simultaneously dampening cell death signaling cascades, as reported for the pro-apoptotic transcription factor p53 [21,22] and the apoptosis signal-regulating kinase 1 (ASK1) [[23], [24], [25]]. In addition to the modulation of signaling cascades, different studies have reported the participation of DJ-1 in the maintenance of mitochondrial homeostasis. Indeed, DJ-1 has been described to interact with complex I [26,27] and, more recently, with the β subunit of F1FO ATP synthase [28], modulating their functionality. Moreover, the protein has been found to take part in the process of mitochondrial quality control, playing a role in the Parkin/PINK1-mediated mitophagy [[29], [30], [31], [32]], and in the regulation of the mitochondrial dynamics [[33], [34], [35], [36]]. Noteworthy, the protein has also been suggested to modulate endoplasmic reticulum-mitochondria tethering, playing a role in the modulation of calcium transients [37,38].
Besides these largely explored functions, more recent findings have suggested that DJ-1 may also exert cellular protection by mitigating dicarbonyl stress. In particular, two glucose-derived metabolites, the methylglyoxal (MGO) and glyoxal (GO), are highly reactive, being able to attack and modify nucleophilic macromolecules, such as proteins and DNA, eventually yielding irreversible advanced glycation end products (AGEs) [39]. In this scenario, DJ-1 has been described to convert MGO and GO in less harmful species, by acting as a glutathione-independent glyoxalase [[40], [41], [42], [43], [44]]. Furthermore, some studies have reported that DJ-1 can deglycate both proteins and nucleic acids upon reaction with MGO [[45], [46], [47], [48], [49], [50]], though this function has proved contentious by other reports [51,52]. Interestingly, DJ-1 also seems to participate in the preservation of glucose homeostasis, by regulating the glucose levels in the brown adipose tissue [53] and by modulating the subcellular localization of the glycolytic enzyme hexokinase I [32]. Moreover, reduced expression levels of DJ-1 have been observed in the pancreatic islets of patients affected by type II diabetes, in contrast to healthy individuals, further supporting the role of the protein in glucose homeostasis [54].
The functions so far described clearly emphasize the protective potential of DJ-1, which appears to play a multifaceted role in orchestrating a wide range of responses to confer cellular protection. Interestingly, through the functions previously described, the protein has also recurrently been involved in the defense against IR injury, a condition that has been correlated with high levels of oxidative stress and underlies several pathologies, ranging from stroke to cancer and diabetes. The pathophysiology of IR injury will be discussed in detail in the following paragraphs, together with the recent studies that have highlighted the participation of DJ-1 in this condition.
Ischemia results from an inadequate blood supply to tissues and organs, commonly due to the obstruction of arterial flow [55]. As a consequence, the body experiences a local shortage of oxygen and nutrients until the blood flow is restored. This condition has relevance not only to stroke, myocardial infarction, and cancer [56] but also to diabetes [57,58], and neurodegenerative pathologies [[59], [60], [61]]. At the molecular level, one of the key events occurring in response to hypoxia is the activation of the transcription factor Hypoxia Inducible Factor 1α (HIF-1α), which otherwise, under normal oxygen tension, is constantly degraded [62]. The oxygen-dependent regulation of HIF-1α is determined by the E3 ubiquitin ligase Von Hippel-Lindau (VHL), which labels HIF-1α for proteasomal degradation. The interaction with VHL is dependent on HIF-1α hydroxylation by the prolyl hydroxylase domain (PHD) protein, which uses oxygen and α-ketoglutarate as substrates [62]. Under limited oxygen availability, HIF-1α is stabilized and orchestrates the expression of multiple genes involved in the hypoxic adaptation, including those participating in vascularization and reprogramming of energy metabolism [63]. Indeed, while under normoxia, ATP synthesis largely relies on oxidative phosphorylation, hypoxic ATP production is driven by glycolysis [55]. Nonetheless, the cellular energetic demand exceeds the glycolytic capacity, eventually leading to a drop in ATP levels. Since ATP is required as a regulator of different ATP-dependent ionic exchangers, the hypoxic period leads to ionic imbalance and cellular acidosis due to the accumulation of lactate [55]. In particular, due to the altered function of Na+/Ca2+ and Na+/H+ exchangers, and of Na+/K+-ATPase, ischemia drives the intracellular accumulation of sodium and calcium, affecting the entire cellular homeostasis (see Fig. 1) [55,64]. Notably, this ionic imbalance becomes even more severe upon reperfusion, when the mitochondrial respiration resumes, and the physiological pH begins to be restored. Indeed, while the extracellular pH is more rapidly normalized with respect to the cytosol, the intracellular compartment remains still partially acidic upon reoxygenation. This condition entails a proton gradient able to sustain the function of the Na+/H+ and Na+/Ca2+ exchangers, further increasing the cytosolic levels of sodium and calcium [65].
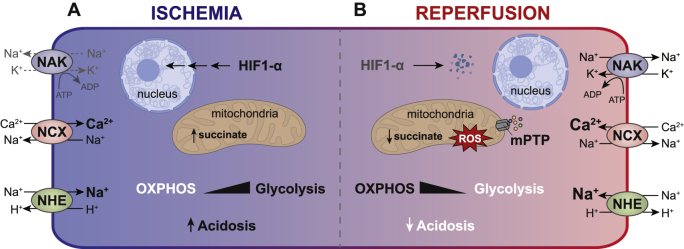
Events contributing to the pathophysiology of IR injury. - (A) Under limited oxygen concentrations, the mitochondrial respiration is inhibited with consequent upregulation of glycolysis and acidosis. To counterbalance the low pH, Na+/H+ exchanger (NHE) exchanges protons for Na+, promoting the build-up of the ion in the cytosol. As Na+/K+ ATPase (NAK) activity is almost inhibited due to the reduced ATP availability, cells exploit the reverse activity of the Na+/Ca+ exchanger (NCX) to counterbalance the increased sodium level. In this way, NCX induces Ca2+ accumulation in the cytosol, while expelling sodium. At the same time, the transcription factor HIF1-α is stabilized and enters the nucleus to promote gene expression. (B) Upon reperfusion, the mitochondrial respiration resumes, re-establishing the normal pH. This rapid pH adjustment causes a large proton gradient that prompts a sustained NHE activity, which additionally rises the cytosolic concentration of sodium. As a consequence, the NCX reverse activity is potentiated, further incrementing the calcium level in the intracellular compartment. In this condition, calcium, in conjunction with the large mitochondrial ROS production, favours the opening of the mPTP, which stimulates the initiation of cell death mechanisms. In this condition, HIF1-α is constantly degraded.
In addition to this ionic dyshomeostasis, the first minutes of reperfusion are characterized by an abundant ROS generation, principally of mitochondrial origin. When the oxygen concentration is reduced, the levels of the mitochondrial metabolite succinate dramatically rise. Succinate is thought to arise due to the reversal activity of complex II, converting fumarate to succinate, though there may also be a contribution from the canonical Krebs cycle supported by glutaminolysis [[66], [67], [68]]. During hypoxia, succinate plays a role in HIF1-α stabilization as it is also transported to the cytosol, where it can inhibit PHDs [69,70]. However, upon reoxygenation, succinate is oxidized back to fumarate by complex II and this event forces electrons to flow from the co-enzyme Q pool back through complex I and onto oxygen through the process referred to as reverse electron transfer (RET), producing large amounts of superoxide anions [66,71]. This initial burst of ROS in conjunction with the conspicuous surge of calcium act as major players in the activation of cell death signaling cascades, which involve the opening of the mitochondrial permeability transition pore (mPTP) [72]. mPTP is considered a high conductance proteinaceous pore localized in the inner mitochondrial membrane [73]. After ischemia, the mPTP opening is stimulated within a few minutes of reperfusion, when the pH is normalized to pre-ischemic values. Indeed, it has been shown that mPTP opening is almost inhibited by pH values below 7.0, probably due to a highly conserved histidyl residue of the mitochondrial F1FO (F)‐ATP synthase complex [74]. Once opened, mPTP increases the permeability of the inner mitochondrial membrane leading to mitochondrial swelling and subsequent activation of cell death pathways [72].
Following the acute phase of reperfusion injury, the release of damage-associated molecular patterns (DAMPs) from injured cells leads to activation of an innate immune response [71,75]. Interestingly, metabolites, such as lactate and succinate, effluxed into the circulation during reperfusion, have also recently been postulated to play a role in this immune activation [76,77]. The initial immune response induces the production of pro-inflammatory cytokines and chemokines, facilitating the infiltration of leukocytes and an inflammatory response [78]. As a consequence, the production of pro-inflammatory cytokines and chemokines facilitate the infiltration of leukocytes and an inflammatory response, which can further exacerbate the extent of tissue damage.
Ischemic injury represents a debilitating pathology and is often regarded as a medical emergency, characterized by its high incidence and a complex clinical picture. Depending on the origin of the ischemic event, the treatment can vary but it commonly comprises the rapid administration of anti-thrombotic, anti-coagulative, and vasodilatory compounds, accompanied by surgical interventions, if necessary [79]. Nonetheless, the intricate etiology of IR injury has rendered it challenging to obtain efficacious therapies to treat the pathology. For this reason, the use of experimental models represents an essential approach to better comprehend this pathological condition and to investigate new therapeutic approaches.
To study ischemia, both in vivo and in vitro models are usually generated by reducing oxygen concentrations or by limiting the utilization of oxygen by organs/tissues. Many animal models rely on the occlusion of the target organ arteries, followed by the release of the suture to reperfuse the tissue [65,80]. Most in vivo studies have been performed in rodents, as their anatomy and physiology display a discrete similarity to humans. Besides in vivo models, researchers also take advantage of in vitro models, in which the selected cellular model is maintained in a hypoxic chamber for the designated experimental time. Moreover, cells can be cultured in a glucose-free acidified medium, which can be supplemented with lactate to mimic its accumulation as normally occurring during anaerobic glycolysis. Successively, reperfusion is promoted by exposing cells to normal oxygen tension and replacing the culture medium with a pH-adjusted one, enriched in essential nutrients [65]. The utilization of experimental models has led to the development of promising therapeutic interventions, including both pharmacological and non-pharmacological treatments. In this regard, hypoxic/ischemic conditioning has emerged as a possible mechanism to lessen the ischemic-associated damage [81]. Conditioning consists of the application of a series of sublethal hypoxic/ischemic cycles, which can be performed before the onset of the ischemic episode to reduce the infarct size (preconditioning) or upon reoxygenation/reperfusion to diminish the associated damage (postconditioning) [[82], [83], [84]]. In addition to pre/postconditioning, remote conditioning has also been reported to display protective effects through the application of brief hypoxic/ischemic episodes to an organ remote from the injured site [85,86]. However, the beneficial effects in human trials are less than clear, with some conflicting results on the protective effects [87]. Furthermore, hypothermia has also been observed to confer protection by lowering the metabolic rate and by delaying the activation of pro-inflammatory and pro-oxidant pathways [[88], [89], [90]]. Besides these non-pharmacological approaches, the explored treatments also include pharmacological strategies, which target molecular pathways known to be affected during hypoxia/ischemia. Thus, the investigated mechanisms encompass the inhibition of Na+/H+ [[91], [92], [93]] and Na+/Ca2+ exchangers to modulate calcium levels [[94], [95], [96]], blockage of mPTP opening [97,98], reduction of cytokine and interleukin release [99,100], and counteracting ROS production [[101], [102], [103]]. Recently, it has become clear that mitochondrial ROS generation importantly contributes to the major damage observed upon hypoxic/ischemic manifestations. Therefore, there is great interest in treatments able to protect against ROS generation, principally targeting mitochondria. In this frame, the reduction of mitochondrial respiration has been observed to reduce the pathology-associated damage [104]. For example, the inhibition of succinate accumulation or prevention of its rapid oxidation upon reoxygenation, by using drugs based on the mitochondrial complex II competitive inhibitor malonate, have proven to display beneficial results [66,101,[105], [106], [107]]. Similarly, blockade of complex I, for example with rotenone [108,109] or preventing complex I reactivation during reperfusion by S-nitrosylation of critical cysteines [[110], [111], [112], [113]], have been demonstrated to ameliorate the detrimental response to reoxygenation. While on the one hand the aforementioned therapeutic approach is to reduce ROS generation, on the other hand there is a focus on upregulating the antioxidant capacity of the cell. The explored approaches include the use of superoxide dismutases and relative mimetics [103,[114], [115], [116]], the mitochondrially targeted antioxidant MitoQ [102,117,118], N-acetylcysteine [119,120], metformin [121,122], which also acts as a weak complex I inhibitor [123], and the administration of natural compounds, such as vitamin C [124]. In addition, recent studies have reported the cardioprotective potential of a mitochondrially targeted MGO and GO scavenger, referred to as MitoGamide, which might be a future promising candidate for IR therapy [125,126]. Besides these compounds in the antioxidant field, interesting insights have been obtained from studies involving the multifaceted protein DJ-1, which is gaining more and more attention as a protective player in response to IR injury.
Compelling evidence has reported the participation of DJ-1 in the response to IR injury. The protein has been described to act on two levels: firstly, by sustaining the hypoxic response under oxygen depletion, and then by protecting against reoxygenation-related damage.
Under limited oxygen concentrations, DJ-1 has been reported to stabilize HIF1-α [127,128]. Vasseur and colleagues initially found that DJ-1 silencing in osteosarcoma-derived cells reduced HIF-1α levels by 30% under hypoxic treatment [128]. Successively, working with neuronal cell models, Parsanejad and co-workers identified VHL as a DJ-1-interacting protein through immunoprecipitation, proposing that DJ-1 may act as a negative regulator of VHL, to boost HIF1-α and prevent its proteasomal degradation [127]. Furthermore, in accordance with other studies [129,130], the authors did not observe any significant reduction in HIF1-α mRNA levels upon DJ-1 deficiency, suggesting that DJ-1 does not influence HIF1-α gene expression [127]. Although the modulation of the VHL–HIF–1α interaction has been proposed as a possible mechanism of action of DJ-1 in neuronal cells, it may not be a general mechanism. Indeed, a more recent study, conducted in colorectal cancer cells, has reported that DJ-1 may promote HIF-1α accumulation through the PI3K/Akt pathway [130], supporting that diverse mechanisms could be activated in different tissues. Moreover, it is relevant to mention that another group has observed HIF-1α protein accumulation upon DJ-1 silencing in neuroblastoma cells [129], suggesting instead, that DJ-1 could play a negative role in HIF1-α stabilization. In an attempt to interpret these contrasting findings, it should be considered that different cell types may express different levels of endogenous DJ-1 and have different susceptibility to the hypoxic treatment. Moreover, the stabilization of HIF-1α is frequently detected via immunoblot, which may yield variable results, considering the labile nature of HIF-1α. Therefore, future analyses are required to clarify how DJ-1 is involved in the regulation of HIF1-α and target genes, and whether the contrasting data reported are due to the diverse experimental model and approach used.
Upon oxygen reintroduction, DJ-1 has been shown to protect against reoxygenation-derived ROS damage, acting at different levels. Indeed, in vivo studies have reported that the absence or down-regulation of DJ-1 results in higher sensitivity to ischemia and increased infarct size in the brain [[131], [132], [133], [134]] and heart [135,136]. On the contrary, DJ-1 upregulation has been observed to elicit neuroprotection and cardioprotection, as its overexpression or injection was reported to rescue post-ischemic cerebral [131,132,137] and myocardial damage [138]. In addition, it is emerging that DJ-1 may play a role in ischemic preconditioning. During in vivo preconditioning, DJ-1 expression levels have been shown to increase, whilst knock-down of the protein seemed to impair the beneficial effects of the treatment [139]. This protective effect has also been observed in cellular models of hypoxic preconditioning, where DJ-1 protein was found to be upregulated [140,141]. Furthermore, two additional studies have emphasized that DJ-1 may play a role during ischemic postconditioning [[142], [143], [144]]. Nonetheless, it is worth mentioning that confounding results have been obtained concerning the modulation of DJ-1 expression levels upon IR. Indeed, while some studies have reported an increased DJ-1 expression [24,[144], [145], [146]], other groups have observed no change [142,147] or even a decreased expression level [148,149]. As these discrepancies may be attributable to different models, experimental procedures, or time windows considered, more studies should be performed to elucidate whether and how DJ-1 expression is transcriptionally regulated under this condition. Moreover, to precisely evaluate its participation in the reperfusion phase, DJ-1 levels should be modulated only during reoxygenation to avoid confounding results deriving from the combination of ischemia and reperfusion injury.
Despite conflicting reports on its expression levels during ischemic events, the protective activity exerted by the protein is becoming more and more evident, even though the precise mechanisms of action remain to be completely elucidated. In this regard, different in vitro reports have found that the protein could act at the mitochondrial level. Accordingly, upon reperfusion, DJ-1 was observed to translocate into mitochondria of rat primary neural cells and isolated murine cardiomyocytes [136,150] and this process might be mediated by the mitochondrial chaperone glucose-regulated protein 75 (Grp75) [142,147]. Moreover, DJ-1 has been described to translocate within the mitochondrial compartment in the hypoxic phase, prior to the reoxygenation process. Indeed, a study reported that the mitochondrial accumulation of DJ-1 under hypoxia occurs in both HEK cells and rat primary cortical neurons and that the translocation may rely on 14-3-3 proteins [151]. In this compartment, the protein has been reported to reduce mitochondrial fragmentation and to sustain complex I activity in cardiac cells [135,136,140,152]. In addition, DJ-1 overexpression has been observed to contribute to a delay in mPTP opening in HL-1 cells subjected to simulated IR [135] and to participate in the protective mechanisms exerted by cyclosporine A, a drug that has been described to prevent mPTP opening [153], supporting a role for DJ-1 in mPTP formation. Therefore, DJ-1 appears to regulate mitochondrial homeostasis through different mechanisms during both ischemia and reperfusion.
Other interesting insights into the involvement of DJ-1 in IR injury have been obtained by a few studies that have highlighted the possible participation of the protein in the modulation of ionic currents. In this frame, a DJ-1-based peptide, named ND-13, has been selected from a library of other DJ-1-related peptides for its ability to exert protection against oxidative and toxic insults in vitro [154]. The peptide has been designed on a conserved portion of DJ-1 (KGAEEMETVIPVD) and attached to a 7 amino-acid region of the cell-penetrating peptide derived from the HIV trans-activator of transcription (TAT) protein (YGRKKRR) [154]. Interestingly, ND-13 has been reported to influence the expression of proteins involved in the activation of Ca2+−activated K+ channels and Kv4 voltage-gated channels, which have been associated with increased resistance to IR injury by reducing the cellular excitability and excitotoxicity, respectively [132]. Moreover, DJ-1 null dopaminergic neurons have been described to display greater hyperpolarization upon oxygen-glucose deprivation than control cells and that this alteration might derive from the participation of DJ-1 in the modulation of the activity of Na+/K+ ATPases, although the underlying mechanisms of action have not been investigated [155]. These studies suggest the possible participation of DJ-1 in the modulation of ion homeostasis during IR, though additional research is required to validate this activity.
Apart from the direct mitochondrial-related functions, other studies have shown that DJ-1 protection could derive from the activation of antioxidant signaling pathways. For example, DJ-1 was demonstrated to induce Nrf2-dependent gene expression, such as SOD2, catalase, and glutathione peroxidase, in rat heart cells [138,156]. In this regard, DJ-1 has been reported to regulate Nrf2 activation also in reactive astrocytes, where the protein is abundantly expressed [133]. Interestingly, the same group has proposed that astrocytic DJ-1 could play an anti-inflammatory role by regulating the expression of inflammatory cytokines and through modulation of the interaction between TRAF6, a member of the TNF receptor-associated factor protein family, and NLRX1, a member of the NOD-like family with anti-inflammatory properties [157]. Additionally, DJ-1 has been described as a mediator of the protective effects from resveratrol against cardiac reoxygenation damage by indirectly driving the deacetylation of the pro-apoptotic transcription factor p53 [146]. Notably, DJ-1 cardioprotection has also been partially attributed to its ability to counteract glycation stress. Indeed, the loss of the protein has been found to favor AGEs accumulation, with consequent activation of AGEs receptors in the murine heart upon IR [158]. In this line, the protein has also been suggested to intervene in the cardioprotection against IR injury in diabetic conditions, possibly by upregulating the Akt cascade and autophagy in diabetic rats subjected to IR injury [143,159,160]. Moreover, the protein seems to elicit a similar protective role in the kidneys, where DJ-1 has been found to be upregulated after myocardial ischemia in diabetic rats, supposing a possible role in this organ [53]. In accordance, DJ-1 overexpression has been reported to defend against oxidative stress in NRK-52E renal cells subjected to hypoxia and high glucose concentrations [161]. Although the function of DJ-1 in the context of diabetic ischemia is still at the dawn, it could represent an interesting aspect to reflect on. As diabetic patients are considered more vulnerable to ischemic damage and present with an increased risk of morbidity [57,162], the ability of DJ-1 to protect against IR injury in hyperglycemic conditions should be further explored.
Besides the ability to act at the cytosolic level, the protein has also been suggested to be protective in the extracellular compartment. Indeed, extracellularly applied DJ-1 has been shown to rescue neuroblastoma cells exposed to oxygen-glucose deprivation, although the precise mechanism remains elusive [163]. In this regard, the secreted form of the protein has been found in oxygen-glucose deprived human neural progenitor cells [150], supporting a role of the protein in the extracellular milieu. Even though the participation of DJ-1 in signaling cascades has been recurrently reported, the picture that emerges from these data renders it difficult to understand whether the pathways here reported represent general targets of DJ-1 activity or whether the protein exerts different functions in a tissue-specific manner. In this frame, it is worth mentioning that while the role of DJ-1 has been mostly examined at the brain level, due to its association with neurodegeneration, less is known about its function in the heart and kidneys or other organs. Therefore, further studies are awaited to examine the role of DJ-1 in these tissues and to clarify whether the protein displays shared or distinct mechanisms of action in different organs.
Although the protective activity of DJ-1 against IR injury is highly supported in the literature, as here discussed, further investigation is required to better comprehend the mechanisms underlying its protection. In this regard, it would be interesting to explore the signals responsible for the activation of the protein. Upon reperfusion, ROS may be responsible for the activation of DJ-1's protective function, suggesting the protein may be redox-sensitive. Therefore, it could be evaluated whether and how the highly conserved Cys-106 residue is involved. Indeed, so far, few studies have addressed this point, mainly reporting that this cysteine is important for DJ-1 to carry out its functions [131,135]. Of note, Ariga's group has performed an in silico virtual screening to find DJ-1-binding compounds that are able to stabilize the reduced and mildly oxidized state of this residue [164]. Interestingly, the results obtained with ‘compounds 23, A and B’ showed the beneficial effects of these molecules in rat models of cerebral ischemia, supporting the therapeutic potential of modulating DJ-1 redox-state [[165], [166], [167]]. Nonetheless, the DJ-1-based peptide, ND-13, which has been shown to improve focal ischemia recovery, does not contain this amino acid, suggesting that this residue may not be the only determinant for its function [132]. Therefore, further research is essential to untangle this aspect. Moreover, while ROS may favor DJ-1 activation upon reoxygenation, under ischemic conditions, where ROS generation is largely repressed due to oxygen depletion, other signals might promote the participation of DJ-1 in HIF-1α stabilization. For example, HIF-1α can be stabilized upon succinate accumulation [69,70]. As DJ-1 has been associated with mitochondrial metabolism [168] and the ND-13 DJ-1-based peptide can regulate succinate dehydrogenase assembly factor 4 protein levels [132], this other mechanism of HIF-1α stabilization should be considered in order to better comprehend the participation of DJ-1 in HIF-1α regulation.
In conclusion, the findings discussed here suggest that the multifaceted behavior of DJ-1 confers the ability to protect against IR injury by orchestrating a wide-spectrum of cellular responses and by acting in multiple tissues (Fig. 2). Although studies robustly validating previously discovered roles, and new studies dissecting the precise mechanisms of action of the protein in this pathological state are essential, DJ-1 may be considered an interesting candidate to investigate for future therapeutic implications in IR injury.
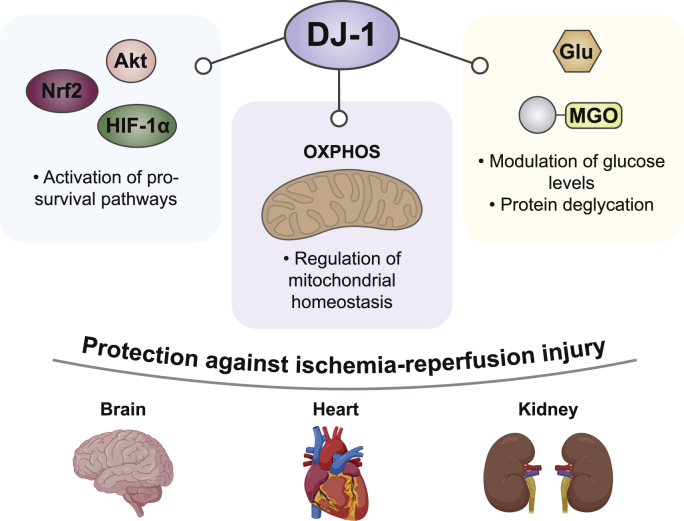
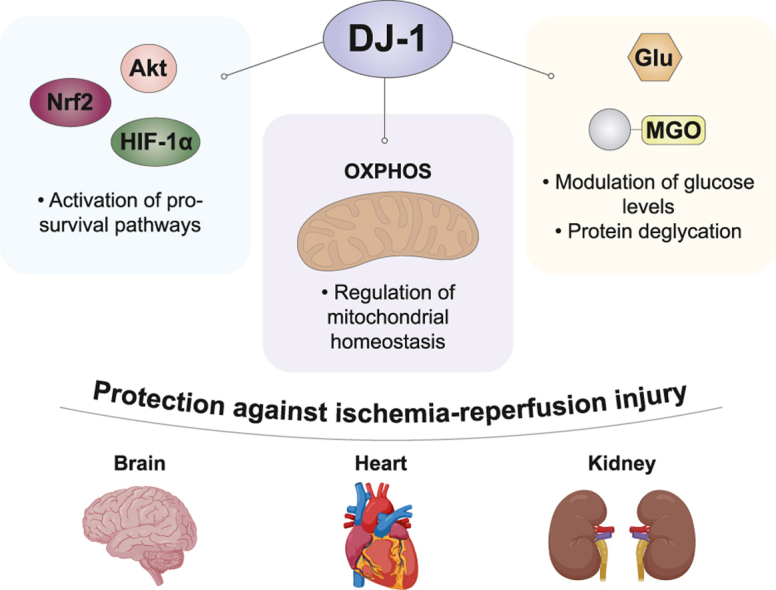
Protective DJ-1 functions under IR injury. - DJ-1 has been reported to provide protection against IR injury through multiple mechanisms. In particular, the protein has been described to sustain adaptive and pro-survival signaling pathways, such as HIF-1α stabilization and Akt and Nrf2 activation, to protect the mitochondrial homeostasis, supporting the normal organelle functionality, and to ameliorate glycation stress, especially in diabetic ischemia. These protective functions have been observed at brain, cardiac and renal level.
The authors declare no competing interests.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
| AGEs | (advanced glycation end products) |
| Akt | (protein kinase B) |
| ASK1 | (apoptosis signal-regulating kinase 1) |
| Ca2+ | (calcium) |
| Cys | (cysteine) |
| DAMPs | (damage-associated molecular patterns) |
| ERK1/2 | (extracellular signal-regulated protein kinases 1 and 2) |
| GO | (glyoxal) |
| Grp75 | (chaperone glucose-regulated protein 75) |
| H+ | (proton) |
| HIF-1α | (hypoxia inducible factor 1α) |
| IP3R | (inositol 1,4,5-trisphosphate receptor) |
| IR | (ischemia-reperfusion) |
| K+ | (potassium) |
| MGO | (methylglyoxal) |
| mPTP | (mitochondrial permeability transition pore) |
| Na+ | (sodium) |
| NLRX1 | (NLR Family Member X1) |
| Nrf2 | (nuclear factor erythroid 2-related factor 2) |
| PHD | (prolyl hydroxylase domain) |
| PI3K | (phosphoinositide 3-kinase) |
| RET | (reverse electron transport) |
| ROS | (reactive oxygen species) |
| SOD | (superoxide dismutase enzyme) |
| TAT | (trans-activator of transcription) |
| TRAF6 | (TNF receptor associated factor 6) |
| VDAC | (voltage-dependent anion channel 1) |
| VHL | (Von Hippel-Lindau) |
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
We thank Prof. Michael P. Murphy for the helpful discussion.
Figures were created in Adobe Illustrator with the support of Biorender.com.
 DJ-1: A promising therapeutic candidate for ischemia-reperfusion injury
DJ-1: A promising therapeutic candidate for ischemia-reperfusion injury
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
