
These authors contributed equally to this work.
Oxidative stress can induce covalent disulfide bond formation between protein-protein thiol groups and generate hydroxyl free radicals that damage DNA. HMGB1 is a DNA chaperone and damage-associated molecular pattern molecule. As a redox-sensitive protein, HMGB1 contains three cysteine residues: Cys23, Cys45, and Cys106. In this study, we focused on the relationship between HMGB1 dimerization and DNA stabilization under oxidative stress conditions. HMGB1 dimerization was positively modulated by CuCl2 and H2O2. Mutation of the Cys106 residue blocked dimer formation. Treatment of HEK293T cells with CuCl2 and H2O2 enhanced the oxidative self-dimerization of HMGB1, whereas this dimerization was inhibited in mutant HMGB1C106A cells. Furthermore, we performed a bimolecular fluorescence complementation assay to visualize Cys106 oxidation-induced HMGB1 dimerization in live cells exposed to oxidative stress and were able to reproduce the dimerization effect of HMGB1 in fluorescence resonance energy transfer analysis. Interestingly, dimerized HMGB1 bound to DNA with higher affinity than monomeric HMGB1. Dimerized HMGB1 protected DNA from damage due to hydroxyl free radicals and prevented cell death. In conclusion, dimerized HMGB1 may play a regulatory role in DNA stabilization under oxidative stress.

•
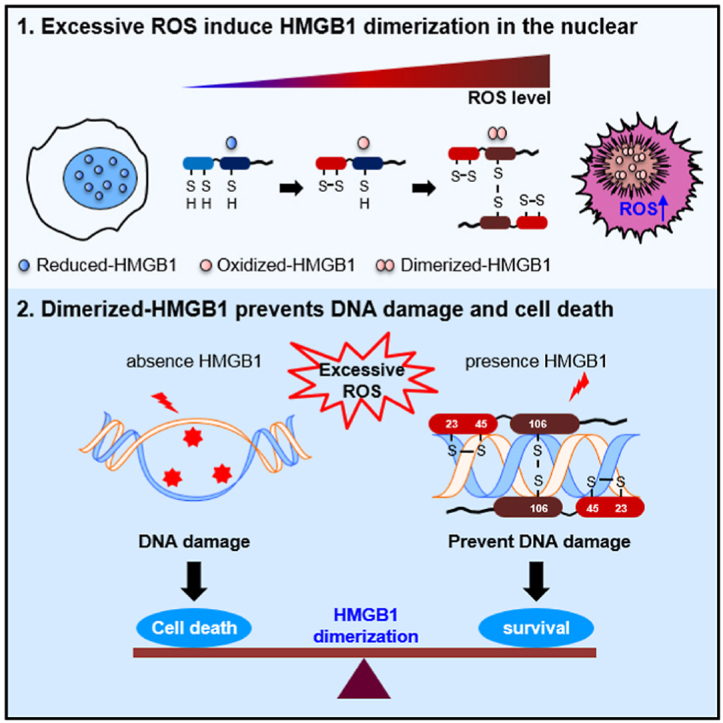
Accumulation of excessive ROS induces DNA damage, causing cell death.
•HMGB1 dimerizes in the presence of excessive ROS and binds DNA with high affinity.
•Binding of dimerized HMGB1 (Di-HMGB1) protects DNA from ROS action.
•We prepared an in vitro HMGB1 dimerization due to excessive ROS.
•Di-HMGB1 protected against DNA damage induced by radiation exposure.
| BiFC | Bimolecular fluorescence complementation |
| CFP | Cyan fluorescent protein |
| Di-HMGB1 | Homo-dimerized HMGB1 |
| FRET | Fluorescence resonance energy transfer |
| GFP | Enhanced green fluorescent protein |
| HMGB1 | High mobility group box1 |
| H2O2 | Hydrogen peroxide |
| LK | Intermediate linker |
| LPS | Lipopolysaccharide |
| MALDI | Matrix-associated laser desorption ionization time-of-flight |
| Ox-HMGB1 | Oxidized HMGB1 between Cys23 and Cys45 |
| PBS | Phosphate-buffered saline |
| PLA | Proximity ligation assay |
| Prx | Peroxiredoxin |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| Re-HMGB1 | Reduced HMGB1 |
| Su-HMGB1 | Sulfonyl HMGB1 |
| STS | Staurosporine |
| WCL | Whole cell lysate |
| WT | Wild-type |
| YFP | Yellow fluorescent protein |
| γ-H2AX | H2AX phosphorylation |
High mobility group box1 (HMGB1) is a highly abundant and ubiquitously expressed nuclear protein that functions as a DNA chaperone and participates in DNA replication, recombination, transcription, and repair [1,2]. It also functions as an extracellular proinflammatory or chemotactic molecule [[3], [4], [5]] when it is actively secreted [[6], [7], [8], [9], [10], [11], [12], [13]] or passively released by necrotic cells, respectively. Extracellular HMGB1 mediates damage-associated molecular pattern signaling by binding to diverse receptors, including the receptor for advanced glycation end products and toll-like receptor-2 and -4 [3,[14], [15], [16], [17], [18]].
HMGB1 is composed of an A box, a B box, and an acidic tail. The A and B boxes bind to the minor groove of DNA and are involved in DNA bending, including V(D)J recombination [19]. HMGB1 contains three cysteines (Cys23 and Cys45 in the A box and Cys106 in the B box) and functions differently depending on its redox state [20]. When all three cysteine residues are in the thiol state, “reduced HMGB1 (Re-HMGB1)” exhibits chemotactic function [21]. When an intramolecular disulfide bond forms among Cys23, Cys45, and Cys106 in the thiol state, “oxidized HMGB1 (Ox-HMGB1)” exhibits a proinflammatory function [22]. When all three cysteines are in the hyperoxidized sulfonic acid state, “sulfonyl HMGB1 (Su-HMGB1)” exhibits no chemotactic or proinflammatory function [23].
Post-translational modifications of HMGB1, such as acetylation [24], phosphorylation [6], and N-glycosylation [25], play critical roles in HMGB1 nucleo-cytoplasmic transport and secretion. In addition, our recent data showed that HMGB1 can be oxidized between Cys23 and Cys45 and translocated into the cytoplasm via thiol peroxidase of peroxiredoxin (Prx) I and II under oxidative stress conditions, and disruption of disulfide bond formation by mutating Cys23 or Cys45 results in almost no secretion of HMGB1 even under oxidative stress conditions [26]. This cytoplasmic translocation and extracellular secretion process also requires Cys106, as evidenced by the cytoplasmic location of mutated Cys106 even in the presence of Cys23 and/or Cys45 mutations [27]. Cytoplasmic Ox-HMGB1 promotes autophagy, playing a crucial role in cell survival during cancer chemotherapy or nutrient depletion. In contrast, extracellular Ox-HMGB1 triggers inflammation [26,[28], [29], [30]]. The Cys106 residue of HMGB1 is important for specific binding to toll-like receptor 4, which induces innate immunity and cytokine release [22].
Reactive oxygen species (ROS) are free radicals that include superoxide anion (O2•-), hydroxyl radical (•OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2). Cellular ROS are mainly generated by mitochondrial nicotinamide adenine dinucleotide phosphate oxidase and are important signaling molecules in many physiological and pathological processes. Intracellular ROS levels regulate both the survival and death of cells [31,32]; lower concentrations of ROS promote cellular proliferation and higher concentrations induce apoptosis or necrotic cell death [[33], [34], [35]]. Copper ions facilitate the formation of ROS, leading to damage to biomolecules such as DNA and chromatin [36]. For example, H2O2 and superoxide, the most reactive and destructive ROS, can cleave the phosphoester bonds between specific nucleotides in DNA. In radiotherapy, ROS formation due to radiation exposure is a well-known primary mechanism of inducing apoptosis in target cancer cells [[37], [38], [39]].
In this study, we evaluated whether HMGB1 could be homo-dimerized at Cys106 in an anti-parallel direction under high oxidative conditions, exceeding the level required for intramolecular disulfide formation both in vitro and in vivo. HMGB1 is a nuclear protein that binds DNA; this binding capacity is even higher for damaged DNA, and Cys106 is a crucial amino acid required for the nuclear localization of HMGB1 [27]. We hypothesized that high intracellular ROS stress conditions induce a nuclear HMGB1 dimer to prevent ROS-mediated DNA damage. Homo-dimerized HMGB1 (Di-HMGB1) exhibits stronger DNA binding than monomeric HMGB1 and protective activity against DNA damage and cell death due to ROS stress.
Wild-type (WT) mouse embryonic fibroblast (MEF), HMGB1-deficient MEF (MEFHmgb1−/-), and HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin and incubated at 37 °C under 5% CO2. For transfection, the cells were plated for 24 h and grown to 80–90% confluency. FuGENE® HD (Promega, Madison, WI, USA) was used for transfection. Electroporation was performed for transfection using MicroPorator-mini (Digital Bio, Korea).
BiFC constructs were generated to observe the homo-dimerization of HMGB1 and the binding orientation. The N-terminal half of enhanced green fluorescent protein (GFP) [amino acids 1–155 (GFPN)] was fused to HMGB1 directly at the 5′ end (GFPN:HMGB1) or indirectly via an intermediate linker (LK) of Ser-(Gly)4-Ser for flexibility (GFPN:LK:HMGB1) [40]. The C-terminal half of GFP [amino acids 156–238 (GFPC)] was fused at the 3′-end of HMGB1 directly (HMGB1:GFPC) or indirectly via the same intermediate LK (HMGB1:LK:GFPC). GFPC fused to the 5′-end of HMGB1 was also cloned (GFPC:HMGB1, GFPC:LK:HMGB1). HEK293T cells were transiently transfected for 32 h at 37 °C and treated with 50 μM CuCl2 and 50 μM H2O2 for 4 h at 37 °C. Transfected cells were observed using a FV1000 confocal microscope (Olympus, Tokyo, Japan). Two repeats of WT HMGB1 or HMGB1C106A were subcloned as GFPN:LK:HMGB1:LK:HMGB1:LK:GFPC [GFPN:LK:(HMGB1)2:LK:GFPC] or GFPN:LK:HMGB1C106A:LK:HMGB1C106A:LK:GFPC [GFPN:LK:(HMGB1C106A)2:LK:GFPC] constructs and transiently overexpressed in HEK293T cells to observe the binding orientation of HMGB1 and the effect of the Cys106 residue of HMGB1.
To determine the function of Di-HMGB1, two repeats of WT HMGB1 or HMGB1C106A, which were linked via LK, were subcloned via insertion into the pCMV-Myc plasmid; these constructs were named Myc-(HMGB1)2 or Myc-(HMGB1C106A)2.
We determined the homo-dimeric binding orientation of HMGB1 by performing a FRET assay with HEK293T cells. YFP:LK:HMGB1, CFP:LK:HMGB1, HMGB1:LK:YFP, and HMGB1:LK:CFP constructs were generated. The YFP:LK:CFP construct was used as a positive control. YFP and CFP are yellow fluorescent protein and cyan fluorescent protein, respectively. Cells were transiently co-transfected with a combination of YFP:LK:HMGB1 and CFP:LK:HMGB1 or YFP:LK:HMGB1 and HMGB1:LK:CFP plasmids. The acceptor photo-bleaching method was used to study HMGB1 homo-dimerization under an FV1000 confocal microscope and using Olympus software. A UPlanAPO 100X NA/1.35 objective was used to observe the binding orientation during the FRET assay. We compared the FRET signal with that of the positive control (YFP:LK:CFP plasmid). Regions of interest were selected, and images were collected before and after bleaching with the 514 nm laser line. The excitation wavelengths were 430 and 514 nm for CFP and YFP, respectively. The two band-pass filters of emission wavelength were 465–510 nm for CFP and 518–561 nm for YFP. A change in the fluorescence intensity between pre- and post-bleach donor values (efficiency, E) was calculated as a percentage according to the following equation: E = [(ICFPpost – ICFPpre)/ICFPpost] × 100.
HMGB1 recombinant protein was purified as described previously [6]. Six His-tagged HMGB1 and HMGB1C106A proteins were produced in Escherichia coli SoluBL21, and HMGB1 boxes A (aa 1–79), B (aa 88–162), and BC106A were produced in E. coli BL21 (DE3) pLysE by adding 0.5 mM isopropyl 1-thio-β-d-galactopyranoside and incubating for 18 h at 24 °C. Recombinant proteins were sequentially processed and purified via Ni2+-NTA, heparin, and gel-filtration column chromatography. Endotoxin was removed using Triton-X114 [41].
To identify the HMGB1 peptide in the HMGB1 dimerization position, whole cell lysates (WCLs) of HEK293T cells were stimulated with H2O2 for 2 h at 37 °C. The HMGB1 dimeric form was separated by non-reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Bands were in-gel digested with trypsin and then run on an LC-MS/MS system through an LTQ-Orbitrap-mass spectrometer (Thermo Electron, Waltham, MA, USA) by ProteomeTech, Inc. (Seoul, Korea).
HMGB1 dimerization was induced via incubation with 10 μM CuCl2 and 100 μM H2O2 for 2 h and at 37 °C. MALDI-TOP MS of HMGB1 dimerization was performed on a Bruker Daltonics Microflex LRF20 instrument (Billerica, MA, USA) equipped with a nitrogen laser (wavelength: 337 nm, pulse repetition rate: 62 Hz; ProteomeTech, Inc.). The examined mass region ranged from m/z 20,000 to 100,000, and the instrument was calibrated externally using a bovine serum albumin standard covering the average masses. The matrix system used for MALDI consisted of sinapinic acid dissolved with 50% acetonitrile and 0.1% trifluoroacetic acid. MALDI mass spectra were acquired from at least six independent spots and accumulated individually from 1000 laser shots for constructing reference spectral profiles.
To observe HMGB1 dimerization, HMGB1, HMGB1C106A, and A and B boxes of HMGB1 proteins were treated with different concentrations of CuCl2 and/or H2O2. The reaction mixture was incubated with phosphate-buffered saline (PBS) containing 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4 (pH 7.4) at 37 °C. To observe HMGB1 dimerization, Western blot analysis was performed under non-denaturing conditions.
For this analysis, SDS-PAGE was performed using Tris-Glycine SDS running buffer or sample buffer not containing β-mercaptoethanol. The blots were transferred to Hybond-ECL nitrocellulose membranes (Amersham plc, Amersham, UK) and processed for immunoblotting. The membranes were incubated with primary antibodies [rabbit anti-HMGB1 (Abcam, Cambridge, UK, ab18256), mouse anti-His (Abcam, ab18184), rabbit anti-pH2AX (H2AX at phosphor S139, Abcam, ab11174), rabbit anti-actin (Cell Signaling Technology, Danvers, MA, USA, 4967), rabbit anti-Flag (Sigma-Aldrich, St. Louis, MO, USA, F7425), mouse anti-Flag (Sigma-Aldrich, F3165), mouse anti-GAPDH (Abfrontier, Seoul, Korea YF-MA10022), rabbit anti-SP1, and mouse anti-Myc antibodies (Cell Signaling Technology, #2276)] for 2 h at room temperature (RT) or overnight at 4 °C. After washing thrice with Tris-buffered saline containing 0.1% Tween 20, the membranes were incubated at RT for 1 h with secondary antibodies conjugated with horseradish peroxidase (Jackson Laboratories, Bar Harbor, ME, USA). The membranes were analyzed using an electrochemiluminescence detection system (GenDEPOT, Katy, TX, USA, W3652-020).
To evaluate HMGB1 homo-dimer formation, a single-molecule pull-down assay was performed as described previously [42]. HEK293T cells were transiently transfected with YFP:LK:HMGB1 and HMGB1:LK:CFP constructs for 36 h. The cells were treated with 50 μM CuCl2/50 μM H2O2 for 4 h and harvested with radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-Cl, pH 7.4, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, and 50 mM NaF) including a protease inhibitor cocktail (GenDEPOT). The WCL was centrifuged at 12,000 rpm for 20 min, and the supernatant was harvested. LabTek™ II chamber (Nunc™, Roskilde, Denmark) was passivated with methoxy polyethylene glycol, and 1 μg of anti-GFP antibody (Abcam, ab5449), which cross-reacts with YFP and CFP, was immobilized for 1 h followed by washing with 0.5% Tween 20 in PBS. The chamber was blocked with 3% bovine serum albumin, and 100 μg WCL was incubated for 16 h at 4 °C. The fluorescence of YFP and CFP was observed under a BX51 fluorescence microscope (Olympus).
To determine the localization of the HMGB1 dimer, HEK293T cells were co-transfected with GFPN:LK:HMGB1 and HMGB1:LK:GFPC plasmids and then treated with 50 μM CuCl2 and 1 mM H2O2 for 4 h to induce excessive oxidative stress. Cells were harvested, and nuclear/cytosolic fractionation was performed using a nuclear/cytosolic fractionation kit (BioVision, Inc., Milpitas, CA, USA, K266) according to the manufacturer's procedure.
To analyze whether the Cys106 residue is important for HMGB1 homo-dimerization, HEK293T and RAW264.7 cells were transiently expressed with Flag- and Myc-tagged HMGB1, HMGB1C106A, or HMGB1C106S plasmids. The cells were treated with 50 μM CuCl2/50 μM H2O2 for 4 h and 10 or 1,000 ng/mL lipopolysaccharide (LPS) for 24 h. The cells were lysed in RIPA buffer containing a protease inhibitor cocktail, and the WCL was centrifuged at 12,000 rpm for 1 min. SureBeads™ magnetic beads tagged with anti-Flag antibody (Bio-Rad, Hercules, CA, USA) were incubated with 200 μg WCL for 18 h at 4 °C. Collected complexes were fractionated via SDS-PAGE after washing, and immunoblotting was performed using anti-Myc antibody.
To examine the DNA-binding affinity of Di-HMGB1, genomic DNA was purified from MEFHmgb1−/- cells using EDTA-free modified RIPA buffer (50 mM Tris, pH 7.4, 1% NP-40, 150 mM NaCl, and 50 mM NaF). HMGB1WT, HMGB1C106A, and Di-HMGB1 proteins were incubated with genomic DNA in WCL, which was isolated from MEFHmgb1−/- cells, under 50 μM H2O2 at 37 °C for 4 h. The mixture was separated on a 0.8% agarose gel and stained with ethidium bromide.
To observe the protective effect of HMGB1 against DNA hydrolysis by DNase I, MEFHmgb1−/- cells were transiently transfected with Myc-HMGB1, Myc-HMGB1C106A, and Myc-(HMGB1)2 plasmids via electroporation for 36 h. WCLs were prepared using EDTA-free modified RIPA buffer. Protein (20 μg) from each WCL was incubated with DNase I to hydrolyze DNA in 10 mM Tris-Cl (pH 7.5) buffer containing 0.25 mM MgCl2 and 10 μM CaCl2 for 10 min at 37 °C. DNase I was inactivated by adding 5 mM EDTA and incubating for 30 min at 50 °C.
To observe the effect of HMGB1 on DNA protection against oxidizing agents, the WCLs prepared above were preincubated with 10 mM H2O2 with or without 10 mM CuCl2 in PBS for 10 min at 37 °C. Total RNA was removed via incubation with RNase A for 30 min at 60 °C. Total proteins in the WCLs were degraded via incubation with Proteinase K containing 2.5 mM EDTA, 10 mM NaCl, and 0.05% SDS for 16 h at 50 °C followed by DNA purification. Genomic DNA was separated on a 0.8% agarose gel and stained with ethidium bromide to observe DNA hydrolysis.
MEFHmgb1−/- cells were transiently transfected with Myc-HMGB1, Myc-HMGB1C106A, and Myc-(HMGB1)2 plasmids for 36 h and then treated 1 μM staurosporine (STS, Sigma-Aldrich) with or without 50 μM Z-VAD for 18 h. To quantify apoptotic cells, the cells were incubated with FITC-annexin V and propidium iodide (BD Biosciences, Franklin Lakes, NJ, USA) for 15 min in the dark. For fluorescence staining of H2AX phosphorylation (γ-H2AX), a marker of DNA damage, MEFHmgb1−/- cells were transfected with the above plasmids and then incubated with 50 μM CuCl2/50 μM H2O2 for 4 h or 50 μM cispaltin for 24 h, or exposed to a radiation dose of 3 Gy for 4 h. The cells were fixed with 4% paraformaldehyde for 20 min at RT and permeabilized with 1% Triton X-100. They were incubated with a blocking agent for 1 h and rabbit anti-pH2AX (Ser 139) antibody (Abcam, ab11175) overnight at 4 °C. After washing, Alexa Fluor 488-conjugated goat anti-rabbit immunoglobulin G antibody (Invitrogen, Carlsbad, CA, USA, A-11008) was added, and the mixture was incubated for 1 h at 37 °C. Stained cells were immediately analyzed via flow cytometry using fluorescence-activated cell sorting BD FACS Verse I (BD Biosciences).
To measure cell viability after inducing oxidative stress, MEFHmgb1−/- cells were transfected with Myc-HMGB1, Myc-HMGB1C106A, and Myc-(HMGB1)2 plasmids and cultured in a 96-well plate. The cells were treated with 1 μM staurosporine (STS) for 24 h, and CCK-8 reagent (Dojindo Molecular Technologies, Kumamoto, Japan, CK04) was added to each well.
Animal studies were performed on age- and gender-matched, 7–8-week-old female C57BL/6 mice. All experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee of Yonsei Laboratory Animal Research Center (YLARC, 2017–0208).
C57BL/6 mice were used to investigate the formation of Di-HMGB1 in tumors produced after irradiation. To generate tumors, 1 × 106 B16F1 cells suspended in PBS were injected into the dorsal subcutaneous area of the mice. After the tumor mass was successfully formed, a set of mice was irradiated with a total of 12 Gy X-ray (4 fractions of 3 Gy radiation) at 3-day intervals, whereas control mice were not exposed to radiation. At 3 h after exposure to the last fraction of irradiation, the mice were sacrificed using CO2. Tumor masses were extracted and gently lysed with 1X RIPA buffer.
BALB/c mice were used to investigate the homo-dimerization of HMGB1 in the serum and damaged-liver. The mice were intraperitoneally injected with 1 mg/kg LPS; after 24 h, serum and liver samples were collected. HMGB1 homo-dimer formation was detected via non-reducing SDS-PAGE.
Homo-dimerization of HMGB1 was evaluated using a Duolink™ II flow PLA Detection Kit (Sigma-Aldrich). RAW264.7 cells were cultured in an eight-well chamber (Nunc™) and transfected with Myc-HMGB1 and Flag-HMGB1 or Flag-HMGB1C106A plasmids. The cells were fixed with 4% paraformaldehyde for 20 min after treatment and blocked for 1 h; mouse monoclonal anti-Myc antibody was added along with rabbit anti-Flag antibody, and the mixture was incubated overnight at 4 °C. After washing, PLA probes were added, and the mixture was incubated for 1 h in a humidity chamber at 37 °C. The samples were treated with a ligation and amplification buffer containing DNA polymerase and fluorescence-labeled oligonucleotides. Fluorescent spots and images were acquired via confocal microscopy.
Experimental data were analyzed via Student's t-test using GraphPad Prism software version 5.0 (GraphPad, Inc., La Jolla, CA, USA). The data represent the mean value and standard error of the mean mentioned in the individual figure legends. Differences were considered statistically significant when the p value was <0.05.
In HMGB1, intramolecular disulfide bonds form between Cys23 and Cys45 with the help of thiol peroxidases PrxI and PrxII under oxidative stress conditions, resulting in the extracellular secretion of HMGB1 [26]. We consistently observed that Ox-HMGB1 formation increased in the presence of H2O2 in a concentration-dependent manner, and complete formation of Ox-HMGB1 was achieved after treatment with 100 μM H2O2 for 2 h or 50 μM H2O2 for 8 h. Unexpectedly, the formation of Di-HMGB1 was observed at a higher concentration of H2O2 (500 μM for 2 h or 50 μM H2O2) after 24 h of treatment (Fig. 1A and B). Copper and iron are ubiquitous metals in living organisms that can cause hydroxyl radical formation. Copper is also associated with tau-related pathology in Alzheimer's disease via the promotion of oxidative stress [43]; we used copper with H2O2 to induce strong oxidative stress in this study. To confirm HMGB1 dimerization through catalytic oxidation reactions in the presence of copper, we confirmed the formation of Di-HMGB1 when the WCL of HEK293T cells was treated with various concentrations of H2O2 and CuCl2 for 2 h (Fig. 1C and D). Di- and Ox-HMGB1 were reduced to monomeric HMGB1 when treated with β-mercaptoethanol (Fig. 1A–D). The Di-HMGB1 band in Fig. 1A was subjected to LC-MS/MS, and the obtained HMGB1 peptide sequence was Leu-Gly-Glu-Met-Trp-Asn-Asn-Thr-Ala-Ala-Asp-Asp-Lys-Gln-Pro-Tyr-Glu-Lys (score: 97), clearly indicating HMGB1 homo-dimer formation. Next, purified monomeric HMGB1 produced from E. coli was treated with 10 μM CuCl2 and 100 μM H2O2 for 2 h and subjected to MALDI-TOF spectrometric analysis (Fig. 1E). We observed Di-HMGB1 at 53.3 kDa and a small portion of the oligomeric form of HMGB1 at around 80 kDa. When HEK293T and MEF cells were treated with 100 μM H2O2 and 10 μM CuCl2 for 16 h, Di-HMGB1 formation was observed in the non-reducing gel (Fig. 1F). Taken together, Re-HMGB1 was oxidized to Ox-HMGB1 at mild ROS concentrations and finally to Di-HMGB1 at excessive ROS concentrations.
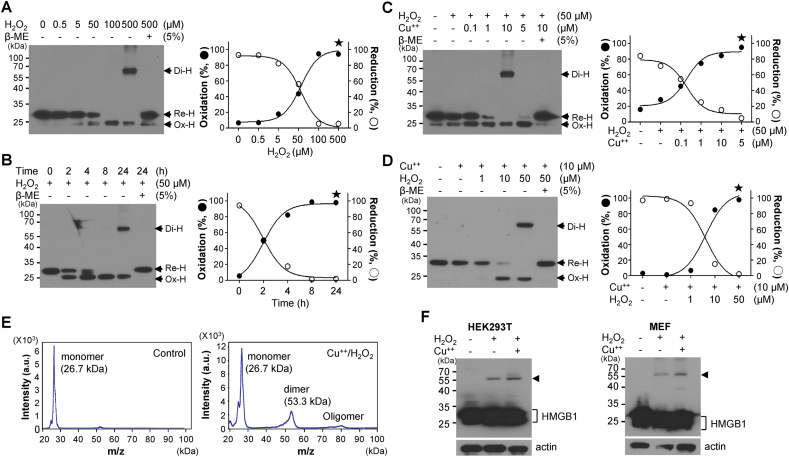
Homo-dimerization of HMGB1. (A, B) Whole cell lysates (WCLs) of HEK293T cells were treated with various concentrations of H2O2 for 2 h (A) or 50 μM H2O2 for various time periods (B) and analyzed via non-reducing SDS-PAGE (left). The percentage change in reduced HMGB1 (Re–H) or oxidized HMGB1 (Ox-H) level was measured (right). Star: dimeric HMGB1 (Di-H). (C, D) HMGB1 was incubated with 50 μM H2O2 and various concentrations of CuCl2 (C) or 10 μM CuCl2 and various concentrations of H2O2 (D) for 2 h at 37 °C and immunoblotted with anti-HMGB1 antibody. β-Mercaptoethanol (5%) was used to reduce HMGB1. (E) MALDI-TOF analysis. HMGB1 was treated with 10 μM CuCl2 and 100 μM H2O2 for 2 h at 37 °C. Control: Buffer treated. (F) HEK293T and MEF cells were treated with H2O2 in the presence or absence of CuCl2 for 14 h, and WCL was analyzed via non-reducing SDS-PAGE.
Among the three cysteine residues, Cys23, Cys45, and Cys106, the HMGB1 motif containing the Cys106 residue is important for the toll-like receptor 4 signaling of HMGB1 [22]. HMGB1 becomes immunologically nonfunctional when the Cys106 residue is hyperoxidized to the sulfonic form (-SO3) [23]. Myc-tagged HMGB1- and HMGB1C106A-transfected WCLs were treated with H2O2/CuCl2 and immunoblotted with anti-Myc antibody. As expected, Di-HMGB1 was observed in only the WT HMGB1-transfected WCLs, whereas Ox-HMGB1 was observed in both HMGB1- and HMGB1C106A-transfected WCLs (Fig. 2A). When recombinant proteins of WT HMGB1 and HMGB1C106A were purified and treated with H2O2/CuCl2, WT HMGB1 formed Di-HMGB1 (Fig. 2B). C106 is located in the B box, and the dimeric or oligomeric form was observed in the only B box when purified A-, BC106A-, and B box proteins were exposed to oxidative stress (Fig. 2C). Interestingly, strong oxidative stress to the HMGB1 A box protein showed oxidation of the A box between Cys23 and Cys45, which migrated upward (Fig. 2C, left panel). Next, HEK293T cells were co-transfected with Flag-HMGB1 and various forms of Myc-HMGB1 plasmids followed by H2O2/CuCl2 treatment; the binding of Flag-HMGB1 and Myc-HMGB1, not HMGB1C106A and HMGB1C106S, was observed upon immunoprecipitation analysis (Fig. 2D). These data indicate that Cys106 is an important residue for HMGB1 dimerization under oxidative stress.
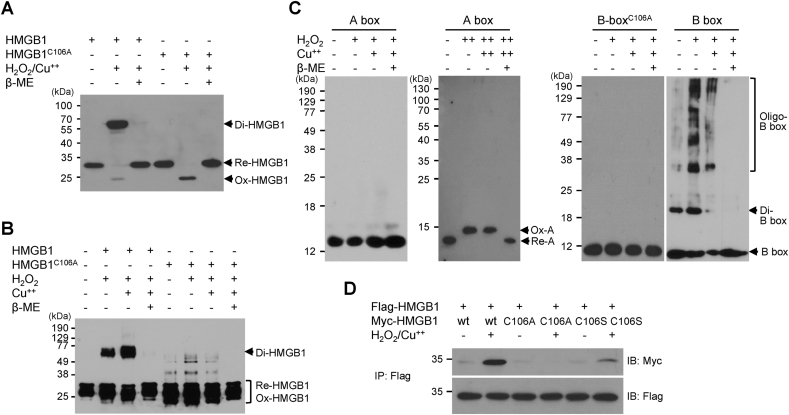
Cys106 is important for HMGB1 homo-dimerization. (A) HEK293T cells were transfected with Myc-tagged HMGB1 and HMGB1C106A, and WCLs were treated with 50 μM H2O2/10 μM CuCl2 in the presence or absence of β-mercaptoethanol. WCLs were immunoblotted with anti-Myc antibody and analyzed via non-reducing SDS-PAGE. (B) HMGB1 and HMGB1C106A protein (100 ng) were incubated with 50 μM H2O2 and 10 μM CuCl2 for 2 h at 37 °C and analyzed via non-reducing SDS-PAGE. (C) HMGB1 A (left panel), BC106A, and B box proteins (100 ng) were incubated with 50 μM H2O2 and 10 μM CuCl2, and HMGB1 A box (right panel) protein was incubated with 100 μM H2O2 and 50 μM CuCl2 in the presence or absence of β-mercaptoethanol for 2 h at 37 °C and immunoblotted with anti-His antibody. The A and B box proteins were subjected to 15% or 12% non-reducing SDS-PAGE, respectively. (D) HEK293T cells were transfected with Flag-HMGB1 or Myc-tagged HMGB1, HMGB1C106A, and HMGB1C106S plasmids for 36 h and treated with 50 μM CuCl2 and 50 μM H2O2. WCLs were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-Myc antibody under denaturing conditions.
Next, we determined whether ROS could promote the formation of Di-HMGB1 in cells. To confirm the binding orientation of Di-HMGB1 (Fig. 3A), BiFC was performed to determine the parallel or anti-parallel orientations of the HMGB1 constructs. Several constructs of HMGB1 were linked to the N- or C-terminal half of GFP (GFPN or GFPC), including the optimal length of the intermediate linker (LK) (Table S1). The BiFC fluorescence signal was observed when both the GFPN:LK:HMGB1 and HMGB1:LK:GFPC constructs were expressed (Fig. 3B, Table S1). This signal was abrogated when HMGB1C106A was used. We developed the GFPN:LK:(HMGB1)2:LK:GFPC plasmid using another method and observed that GFP protein complementation was increased by 5.8-fold after H2O2/CuCl2 treatment of HEK293T cells transfected with this plasmid compared to that in cells transfected with the GFPN:LK:(HMGB1C106A)2:LK:GFPC plasmid. (Fig. 3C). The Di-HMGB1 form was localized in the nucleus (Fig. 3B and C), as confirmed by nuclear fractionation (Suppl. Fig. 1A). Next, we performed a complementary FRET assay after photo-bleaching to show the dimeric binding of HMGB1. HEK293T cells were transfected with the YFP:LK:HMGB1, CFP:LK:HMGB1, and HMGB1:LK:CFP constructs. The YFP:LK:CFP construct was used as a positive control. Increased donor fluorescence after photo-bleaching indicates energy transfer between the donor and recipient, which can only occur when the donor-recipient distance is less than 10 nm [44]. FRET signals of CFP to YFP were obtained before and after photo-bleaching. The FRET value of the anti-parallel orientation of the HMGB1 constructs obtained using YFP:LK:HMGB1 and HMGB1:LK:CFP was 17.13 ± 4.9%, whereas the FRET value of the parallel orientation of YFP:LK:HMGB1 with CFP:LK:HMGB1 was 10.58 ± 1.53% (Fig. 3D). As cellular protein complexes were probed by performing a single-molecule pull-down assay [42], we tested Di-HMGB1 formation using the YFP:LK:HMGB1 and HMGB1:LK:CFP constructs. After HEK293T was transfected with YFP:LK:HMGB1 and HMGB1:LK:CFP, we observed the co-localization of YFP and CFP fluorescence, with a significantly increased signal intensity (by 2.78-fold) compared to the control (Suppl. Fig. 1B). These results indicate that direct visualization of Cys106-mediated HMGB1 homo-dimerization in the anti-parallel orientation is increased because of ROS in living cells.
![HMGB1 homo-dimerization in the anti-parallel direction. (A) Hypothetical model of HMGB1 dimerization. (B, C) Schematic HMGB1 constructs and dimeric HMGB1 constructs for the BiFC assay. GFPN, GFPC: N- or C-terminal half of GFP. LK: linker. HEK293T cells were transfected with a mixture of GFPN:LK:HMGB1 and HMGB1:LK:GFPC, GFPN:LK:HMGB1C106A and HMGB1:LK:GFPC, or GFPN:LK:HMGB1 and HMGB1 plasmids (B) and transfected with GFPN:LK:(HMGB1)2:LK:GFPC and GFPN:LK:(HMGB1C106A)2:LK:GFPC plasmids (C) for 36 h and treated with 50 μM CuCl2 and 50 μM H2O2 for 4 h. The cells were fixed and observed under a confocal microscope. Fluorescence intensities of signal-positive cells were calculated using FV1000 software, and all data are expressed as the means ± SEM (n = 3, right panel). **p < 0.001, t-test. (D) FRET analysis by acceptor photo-bleaching. HEK293T cells were transfected with a mixture of YFP:LK:HMGB1 and CFP:LK:HMGB1 or YFP:LK:HMGB1 and HMGB1:LK:CFP plasmids. The YFP:LK:CFP plasmid was used as control. Cells were cultured in coverslip chambers precoated with poly-L-Lys for 36 h and observed under a confocal microscope. FRET efficiency was determined in a higher fluorescence area and calculated in percentage as E = [(ICFPpost – ICFPpre)/ICFPpost] × 100 (right panel). FRET efficiency data were calculated from three independent experiments using at least 10 images from each sample. All data are expressed as the means ± SEM. *p < 0.05, **p < 0.001, t-test.](/dataresources/secured/content-1765799864991-64eb77b8-c897-4c0e-9be6-1792730cfdbc/assets/gr3.jpg)
HMGB1 homo-dimerization in the anti-parallel direction. (A) Hypothetical model of HMGB1 dimerization. (B, C) Schematic HMGB1 constructs and dimeric HMGB1 constructs for the BiFC assay. GFPN, GFPC: N- or C-terminal half of GFP. LK: linker. HEK293T cells were transfected with a mixture of GFPN:LK:HMGB1 and HMGB1:LK:GFPC, GFPN:LK:HMGB1C106A and HMGB1:LK:GFPC, or GFPN:LK:HMGB1 and HMGB1 plasmids (B) and transfected with GFPN:LK:(HMGB1)2:LK:GFPC and GFPN:LK:(HMGB1C106A)2:LK:GFPC plasmids (C) for 36 h and treated with 50 μM CuCl2 and 50 μM H2O2 for 4 h. The cells were fixed and observed under a confocal microscope. Fluorescence intensities of signal-positive cells were calculated using FV1000 software, and all data are expressed as the means ± SEM (n = 3, right panel). **p < 0.001, t-test. (D) FRET analysis by acceptor photo-bleaching. HEK293T cells were transfected with a mixture of YFP:LK:HMGB1 and CFP:LK:HMGB1 or YFP:LK:HMGB1 and HMGB1:LK:CFP plasmids. The YFP:LK:CFP plasmid was used as control. Cells were cultured in coverslip chambers precoated with poly-L-Lys for 36 h and observed under a confocal microscope. FRET efficiency was determined in a higher fluorescence area and calculated in percentage as E = [(ICFPpost – ICFPpre)/ICFPpost] × 100 (right panel). FRET efficiency data were calculated from three independent experiments using at least 10 images from each sample. All data are expressed as the means ± SEM. *p < 0.05, **p < 0.001, t-test.
The interaction between HMGB1 and chromatin can be measured by performing DNA-binding affinity and DNA stability assays [[45], [46], [47]]. We next investigated the role of Di-HMGB in cells. HMGB1 molecules were incubated in 37 °C for 4 h in PBS containing 50 μM H2O2 and 50 μM CuCl2 to obtain Di-HMGB1. Approximately 70% of the HMGB1 molecules were dimers, whereas a small fraction existed in the oligomeric form (Fig. 4A). The interaction between HMGB1 and genomic DNA in MEFHmgb1−/- cells was measured using the electrophoretic mobility shift assay. As expected, the migration of genomic DNA incubated with Di-HMGB1 was inhibited in the presence of HMGB1 in a dose-dependent manner compared to that in the presence of monomeric HMGB1 (Fig. 4B and C). Genomic DNA, which was incubated with HMGB1, showed slow migration in the presence of 50 μM H2O2 compared to that in the presence of HMGB1C106A (Fig. 4D and E), suggesting that HMGB1 dimerization occurred under oxidative stress and that the dimeric form exhibited greater DNA-binding affinity. Next, the protective effect of Di-HMGB1 against DNA damage was confirmed. We determined the protective function of Di-HMGB1 under strong ROS conditions by performing a DNA protection assay using DNase I. MEF cells exhibited a superior DNA protective effect compared to MEFHmgb1−/- cells against DNase I in dose- and time-dependent manners (Fig. 4F and G). To investigate whether HMGB1 directly protected DNA from the activity of DNase I, the binding of Di-HMGB1 to DNA was tested. Genomic DNA from MEFHmgb1−/- cells was mixed with monomeric or Di-HMGB1 and treated with different units of DNase I. Genomic DNA was dose-dependently hydrolyzed by DNase I and completely hydrolyzed with 1 unit of DNase I in the absence of HMGB1. However, DNA hydrolysis was inhibited by 2 μg/mL HMGB1 at 1 unit of DNase I, and this inhibition was stronger when Di-HMGB1 was added (Fig. 4H). Next, MEFHmgb1−/- cells were transfected with Myc-HMGB1 and Myc-(HMGB1)2, and WCLs were incubated with different concentrations of DNase I. The DNA protective effect in Myc-(HMGB1)2-transfected cells was greater than that in those transfected with Myc-HMGB1 or a vector (Fig. 4I). Taken together, Cys106-mediated Di-HMGB1 preferentially binds to genomic DNA and effectively prevents DNA damage.
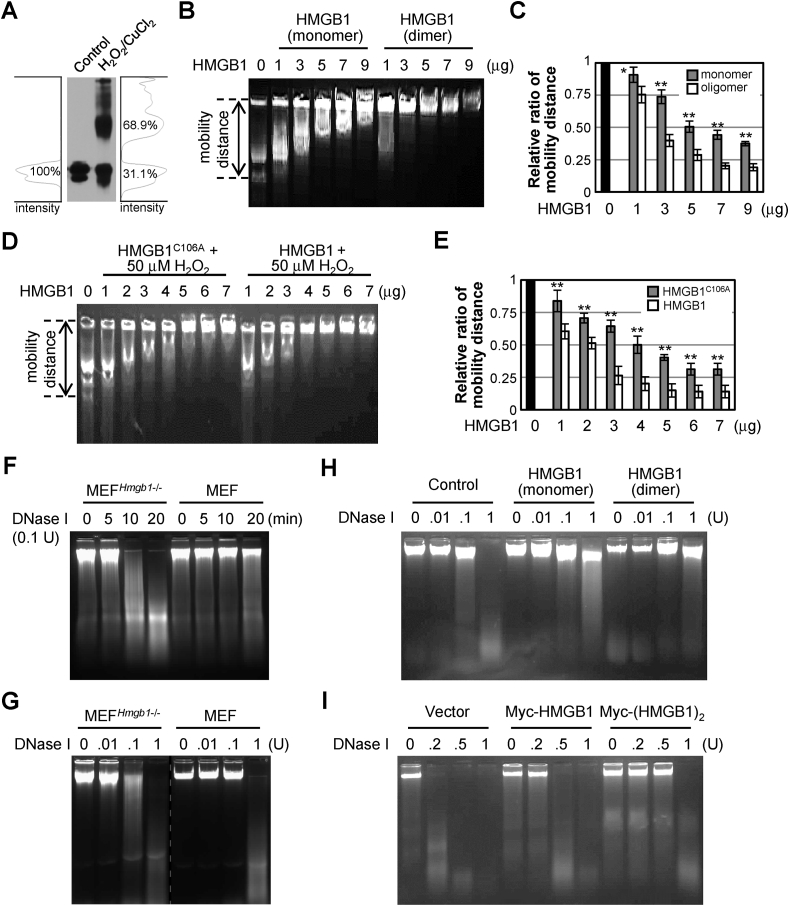
Di-HMGB1 is important for DNA binding and protection of DNA from DNase I action. (A) Dimeric form of HMGB1 produced via incubation with 50 μM CuCl2 and 50 μM H2O2 for 4 h at 37 °C. (B, C) WCL containing genomic DNA from MEFHmgb1−/- cells was incubated with various amounts of monomeric or a mixture of monomeric and dimeric HMGB1 (B), and the migration distances were measured from each well to the DNA tail for the relative ratio of mobility distance (C). (D, E) WCL containing genomic DNA from MEFHmgb1−/- cells was incubated with various amounts of HMGB1 or HMGB1C106A under 50 μM H2O2 for 4 h, and the migration distances were measured (E). All data (C, E) are expressed as the means ± SEM (n = 3). *p < 0.01, **p < 0.001, t-test. (F, G) WCLs of MEF and MEFHmgb1−/- cells were incubated with 0.1 unit DNase I for various time periods (F) or with different amounts of DNase I (G) for 5 min at 37 °C. EDTA at 5 mM was used to stop the DNase I reaction, and aliquots were separated on a 0.8% agarose gel. (H) WCL containing genomic DNA from MEFHmgb1−/- cells was incubated with various amounts of DNase I in the presence of monomeric or dimeric HMGB1 for DNA hydrolysis at 37 °C for 5 min. (I) MEFHmgb1−/- cells were transiently overexpressed with Myc-HMGB1 or Myc-(HMGB1)2 plasmid for 36 h, and each WCL was incubated with various units of DNase I. (F–I) All experiments were repeated three times and showed similar results as the representative data shown.
HMGB1 binds to the minor groove of DNA and its binding affinity increases for UV-induced DNA damage or cisplatin-modified DNA [[48], [49], [50]]. Hydroxyl free radicals primarily cause DNA damage [51]. We hypothesized that Di-HMGB1 is more capable of protecting DNA from damage caused by hydroxyl free radicals than monomeric HMGB1 or blank control. To confirm this hypothesis, MEFHmgb1−/- cells were transfected with a construct of Myc-HMGB1 or Myc-(HMGB1)2 plasmid for 36 h and then incubated with H2O2/CuCl2 for 4 h. Genomic DNA was completely degraded after H2O2/CuCl2 treatment in mock-transfected cells but Myc-(HMGB1)2 transfection showed profound protection of genomic DNA from ROS-induced damage (Suppl. Fig. 2A). We tested the phosphorylation level of histone H2AX, which increased against the DNA damage caused by ROS [52]. MEFHmgb1−/- cells transfected with the Myc-(HMGB1)2 plasmid for 36 h showed the most dramatic decrease in γ-H2AX levels. Myc-HMGB1 overexpression decreased γ-H2AX levels to a greater extent than that observed after treatment with an empty vector. In contrast, Myc-(HMGB1C106A)2 overexpression showed a high level of γ-H2AX, similar to that observed in the vector control (Fig. 5A–C). Ionizing radiation induces double-strand breaks in DNA by generating free radicals [53]. In the irradiation analysis (3 Gy for 4 h), the change in the γ-H2AX level showed a similar pattern (Fig. 5D–F). When we used cisplatin, a DNA-damaging agent, HMGB1 and (HMGB1)2 reduced the γ-H2AX level but Myc-(HMGB1C106A)2 treatment produced almost no such effect (Suppl. Fig. 2B). These results demonstrate that Cys106-mediated Di-HMGB1 prevents ROS-induced DNA hydrolysis.
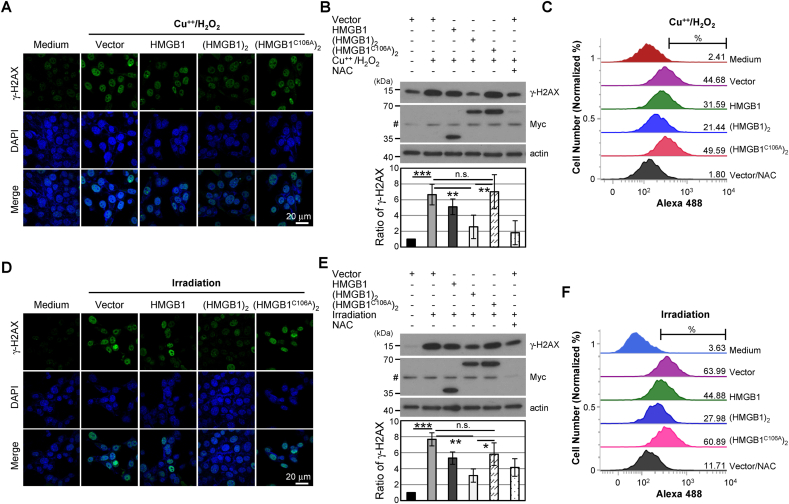
Di-HMGB1 effectively decreases ROS-mediated DNA damage. MEFHmgb1−/- cells were transfected with vector control (vector), wild-type Myc-HMGB1 and Myc-(HMGB1)2, and Myc-(HMGB1C106A)2 plasmid and treated with 50 μM CuCl2/50 μM H2O2 for 4 h (A–C) or with irradiation (3 Gy) for 4 h (D–F). The cells were immunostained against γ-H2AX for confocal microscopy (A, D), and Western blot analysis (B, E) and flow cytometry analyses were performed (C, F). Bar: 20 μm, n.s.: not significant, *p < 0.05, **p < 0.01, ***p < 0.001, t-test.
To determine whether Di-HMGB1 protects cells from ROS-mediated apoptosis, MEFHmgb1−/- cells were transiently transfected with several types of HMGB1 plasmids and then treated with STS, a well-known apoptosis inducer in almost all cell lines. Flow cytometric analysis showed that the percentage of apoptotic cells increased in mock plasmid-transfected cells after STS treatment but Myc-(HMGB1)2 plasmid-overexpressing cells were protected against STS-induced apoptosis (Suppl. Fig. 3A). We used Z-VAD, a pan-caspase inhibitor, for apoptosis inhibition. According to the results of the CCK assay, the viability of Myc-(HMGB1)2-transfected cells was increased compared to that in those transfected with the vector control (Suppl. Fig. 3B). Similar results were obtained after H2O2 treatment (Fig. S3C). MEFHmgb1−/- cells were more susceptible to STS-induced apoptosis than MEF cells (Fig. S3D and E). These results indicate that Cys106-mediated Di-HMGB1 prevents apoptotic cell death.
To determine the physiological relevance of HMGB1 dimerization, MEFHmgb1−/- cells were transfected with Myc-tagged HMGB1 and Flag-tagged HMGB1 plasmids and then treated with low and high concentrations of LPS (10 and 1,000 ng/mL), respectively. Dimer formation in Flag-HMGB1- and Myc-HMGB1-transfected cells other than HMGB1C106A was observed via immunoprecipitation analysis and PLA assay after treatment with a high concentration of LPS (Fig. 6A and B).
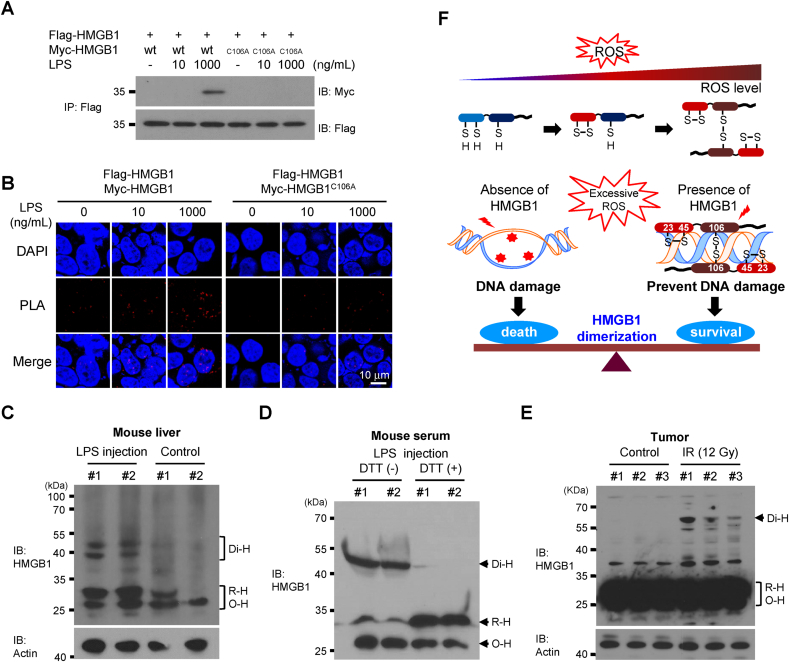
Di-HMGB1 formation in LPS-treated MEF cells and endotoxemia mouse model. MEFHmgb1−/- cells were transfected with wild-type Flag-HMGB1 and Myc-tagged HMGB1 or HMGB1C106A plasmids for 36 h and treated with 10 or 1,000 ng/mL LPS. WCLs were immunoprecipitated with anti-Flag Ab and immunoblotted with anti-Myc antibody (A), or PLA was analyzed with rabbit anti-Flag antibody and mouse anti-Myc antibody (B). Two BALB/c mice were intraperitoneally injected with 1 mg/kg LPS. Liver tissues were collected and minced, and 5 μg of WCL was immunoblotted with anti-HMGB1 antibody (C). Blood and liver tissues were serially obtained after 24 h. Blood serum (10 μL) was separated by non-reducing SDS-PAGE in the presence or absence of 5 mM dithiothreitol and immunoblotted with anti-HMGB1 Ab (D). B16F1 cells were injected into the dorsal subcutaneous area of C57BL/6 mice, and 12 Gy X-ray were irradiated at 3-day intervals with 4 fractions of 3 Gy radiation. Tumor masses were extracted, gently lysed, and analyzed by non-reducing SDS-PAGE (E). (F) A model of the HMGB1 homo-dimerization process depending on the ROS concentration. HMGB1 homo-dimerized under excessive ROS conditions and protected DNA against ROS-mediated damage.
Next, we injected BALB/c mice with 1 mg/kg of LPS to observe the formation of Di-HMGB1 under oxidative stress conditions in vivo. ROS is one of main mediators of LPS-stimulated signaling [54]. To identify ROS-induced HMGB1 dimerization, we obtained mouse liver and serum samples after LPS injection for non-reducing Western blot analysis. LPS-mediated HMGB1 dimerization was detected in the liver samples of the LPS injection group but dimerization was almost negligible in the liver samples from the PBS-treated control group (Fig. 6C). Moreover, the detection of Di-HMGB1 was confirmed in the serum samples of LPS-injected mice by treating the samples with dithiothreitol to reduce Di-HMGB1 to its monomeric form (Fig. 6D). In addition, we investigated HMGB1 dimerization in tumors induced in C57BL/6 mice exposed to therapeutic irradiation. We confirmed HMGB1 dimerization in the irradiation treatment group but not in the control group (Fig. 6E).
In summary, we prepared an in vitro model of HMGB1 dimer formation due to excessive ROS and evaluated its preventative role in DNA damage and cell death by ROS assault (Fig. 6F). ROS significantly accumulate under abiotic and biotic stress conditions, and the accumulation of excessive ROS induces DNA damage and subsequent cell death. HMGB1 is a ubiquitous nuclear protein expressed in all nucleated cells and platelets; an intramolecular disulfide bond is formed under mild oxidative stress conditions and homo-dimers are formed in the presence of excessive ROS. Di-HMGB1 exhibits increased DNA-binding affinity, which can protect DNA from ROS-mediated damage.
HMGB1 is a redox-sensitive molecule that converts from the reduced form to its disulfide form via an intramolecular bond between the Cys23 and Cys45 residues under oxidative stress [26,27]. When RAW264.7 and MEF cells are stimulated, reduced HMGB1 in the nucleus is oxidized to form disulfide HMGB1 by PrxI/II peroxidase and then translocated to the cytoplasm to be secreted as an extracellular proinflammatory molecule [26]. Our study showed that Di-HMGB1 was formed via the Cys106 residue in an anti-parallel direction after intramolecular disulfide formation as the H2O2 concentration increased.
In this study, we found that HMGB1 was dimerized under high ROS conditions in vitro and in vivo, whereas low ROS concentrations caused the formation of intramolecular disulfide bonds in HMGB1 via PrxI/II peroxidase. Di-HMGB1 exhibited increased binding to genomic DNA and protected it from the degradative effects of oxidative stress, whereas HMGB1C106A exhibited almost no dimer formation, reducing DNA protection against ROS assault. However, the underlying mechanism of the DNA-protecting effect of Di-HMGB1 is unclear. HMGB1 is abundant in the nucleus, and its intramolecular (Cys23-Cys45) and intermolecular oxidation (Cys106) may result from the ROS removal process, which allows Di-HMGB1 to strongly bind to genomic DNA to prevent the DNA damage caused by hydroxyl free radicals. Our data support that the transient overexpression of WT HMGB1 decreases the γ-H2AX level after H2O2 treatment and irradiation rather than C106A HMGB1 overexpression, indicating cell protection due to WT HMGB1 after H2O2 treatment and irradiation. Moreover, the γ-H2AX level after H2O2 treatment and irradiation was lower in Di-HMGB1-overexpressing cells than in WT HMGB1-expressing cells. Our findings are consistent with those of previous studies showing that chemical cross-linker- or DsRed-mediated HMGB1 oligomerization increases the binding affinity and stability of HMGB1 on chromatin [45,47].
Di-HMGB1 was resistant to the denaturation caused by SDS treatment and boiling (data not shown) but was susceptible to the denaturation caused by β-mercaptoethanol, indicating that HMGB1 dimerization is caused by cysteine-mediated oxidation. Furthermore, C106A HMGB1 remains in its monomeric form under high ROS conditions, and the Cys106-mediated dimerization of HMGB1 occurs only under excessive oxidative stress, such as that induced by LPS injection. However, how HMGB1 molecules aggregate to oxidize the Cys106 residue and produce the dimeric form is unclear. Previously, the concept of HMGB1 dimerization or oligomerization as a regulatory mechanism of DNA chaperones was tested using artificial integrators such as DsRed or a chemical cross-linker [45,47]. DsRed-mediated oligomerization of HMGB1 can function as a DNA chaperone, as it increases the stability of HMGB1 on chromatin [47]. Here, we physiologically observed the formation of Di-HMGB1 after irradiation in the LPS-induced endotoxemia model. The binding of Di-HMGB1 with chromatin prevents DNA damage due to oxidative stress. Cells exposed to ROS eventually die because of DNA hydrolysis. The protection of DNA against ROS-mediated hydrolysis indicates a role for Di-HMGB1 in cell survival. In contrast, the downregulation of endogenous HMGB1 by short hairpin RNA has been shown to inhibit cell survival in lymph node carcinoma of prostate cancer cells [55]. We found a considerable amount of Di-HMGB1 in the serum of the LPS-induced endotoxemia model mice. It is not known how extracellular Di-HMGB1 is formed, as Di-HMGB1 is mainly located in the nucleus. Serum Di-HMGB1 appears to form from monomeric HMGB1 after serum ROS levels, which are increased by LPS injection [56]. The passive release of Di-HMGB1 after cell damage to the extracellular space is possible. The functional significance of Di-HMGB1 in the blood must be further investigated. Extracellular Di-HMGB1 induced a greater proinflammatory response than HMGB1 monomer (data not shown), and the effects of Di-HMGB1 on the microenvironment, including inflammation and apoptosis, require additional analysis. Previous studies showed that increased oxidation converts Re-HMGB1 to Ox-HMGB1 between Cys23 and Cys45 to Su-HMGB1 or hyperoxidized HMGB1, which is a non-inflammatory molecule [23,57]. We could not detect Su-HMGB1 under oxidative conditions using our LC-MS/MS system [26], and further in-depth study is necessary to determine whether Su-HMGB1 exists and to show how dimeric and sulfonyl HMGB1 are produced and influence immunological function.
To investigate the role of Di-HMGB1 in preventing DNA damage due to peroxidation, we used WCLs, including genomic DNA from MEFHmgb1−/- cells cultured in EDTA-free modified RIPA buffer. DNase I cleaves the phosphodiester backbone of the DNA double helix by hydrolyzing the P–O3′-bond, yielding 5′-phosphorylated fragments [58,59]. We observed that Di-HMGB1 strongly bound to DNA and prevented genomic DNA damage due to hydrolysis by DNase I and H2O2 with or without CuCl2. Therefore, the Cys106-mediated dimerization of HMGB1 in the presence of excessive ROS plays a key role in cell survival by protecting against peroxidation-induced DNA damage.
In conclusion, we demonstrated that Cys106-mediated HMGB1 dimerization occurs in the presence of high ROS concentrations, and the formed dimers strongly bind to DNA to prevent its hydrolysis in the presence of excessive ROS (such as that following radiotherapy for cancer) and eventually increase cell survival.
The authors declare that they have no conflict of interest.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
None.
This study was supported by grants from the
None.
 Reactive oxygen species induce Cys106-mediated anti-parallel HMGB1 dimerization that protects against DNA damage
Reactive oxygen species induce Cys106-mediated anti-parallel HMGB1 dimerization that protects against DNA damage
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
