
Equal author contributions.
During pregnancy, estrogen (E2) stimulates uterine artery blood flow (UBF) by enhancing nitric oxide (NO)-dependent vasodilation. Cystathionine γ-lyase (CSE) promotes vascular NO signaling by producing hydrogen sulfide (H2S) and by maintaining the ratio of reduced-to-oxidized intracellular glutathione (GSH/GSSG) through l-cysteine production. Because redox homeostasis can influence NO signaling, we hypothesized that CSE mediates E2 stimulation of UBF by modulating local intracellular cysteine metabolism and GSH/GSSG levels to promote redox homeostasis. Using non-pregnant ovariectomized WT and CSE-null (CSE KO) mice, we performed micro-ultrasound of mouse uterine and renal arteries to assess changes in blood flow upon exogenous E2 stimulation. We quantified serum and uterine artery NO metabolites (NOx), serum amino acids, and uterine and renal artery GSH/GSSG. WT and CSE KO mice exhibited similar baseline uterine and renal blood flow. Unlike WT, CSE KO mice did not exhibit expected E2 stimulation of UBF. Renal blood flow was E2-insensitive for both genotypes. While serum and uterine artery NOx were similar between genotypes at baseline, E2 decreased NOx in CSE KO serum. Cysteine was also lower in CSE KO serum, while citrulline and homocysteine levels were elevated. E2 and CSE deletion additively decreased GSH/GSSG in uterine arteries. In contrast, renal artery GSH/GSSG was insensitive to E2 or CSE deletion. Together, these findings suggest that CSE maintenance of uterine artery GSH/GSSG facilitates nitrergic signaling in uterine arteries and is required for normal E2 stimulation of UBF. These data have implications for pregnancy pathophysiology and the selective hormone responses of specific vascular beds.

•
CSE-null mice exhibit abnormal estrogen augmentation of uterine artery blood flow.
•Estrogen lowers uterine artery nitric oxide metabolites in CSE null mice.
•CSE loss and estrogen additively impair uterine artery glutathione homeostasis.
•Neither CSE loss nor estrogen influences renal artery blood flow or glutathione.
| CBS | cystathionine β-synthase |
| cGMP | cyclic guanosine monophosphate |
| CSE | cystathionine γ-lyase |
| CSE KO | CSE-null mice. |
| CV | coefficient of variation |
| E2 | estrogen (17β-estradiol) |
| EDV | end diastolic velocity |
| eNOS | endothelial nitric oxide synthase |
| GSH | reduced glutathione |
| GSSG | glutathione disulfide. |
| H2S | hydrogen sulfide. |
| NO | nitric oxide. |
| NOS | nitric oxide synthase |
| NOx | nitrate and nitrite |
| P4 | progesterone |
| PDE5 | phosphodiesterase 5 |
| PSV | peak systolic velocity |
| PI | pulsatility index |
| PKG | cGMP-dependent protein kinase G |
| RI | resistance index |
| sGC | soluble guanylate cyclase |
| UBF | uterine blood flow. WT, wildtype |
Estrogen (17β-estradiol; E2) regulates uterine artery blood flow (UBF) via the gasotransmitter nitric oxide (NO) [1]. Following menses and during pregnancy, rising E2 levels activate endothelial NO synthase (eNOS) to produce NO from arginine [1,2]. NO stimulates the soluble guanylate cyclase (sGC)-protein kinase G (PKG) pathway, which promotes relaxation of vascular myocytes and causes vasodilation and increased blood flow [[3], [4], [5], [6], [7]]. E2-NO regulation of UBF is also sensitive to progesterone (P4), which can induce both relaxation and contraction of uterine arteries [8,9]. In addition to its vasodilatory role, E2 influences redox homeostasis. E2 attenuates pro-oxidant NFκB and TNFα pathways [10,11], but paradoxically increases superoxide (O2−) and hydrogen peroxide (H2O2) production [12,13]. In some situations, E2 replacement therapy can exacerbate post-menopausal cardiovascular disease [14]. This may be due to oxidative stress promoting eNOS uncoupling to favor O2− synthesis and NO oxidation to cytotoxic peroxynitrite [1,14,15]. It is unclear whether E2 affects the uterine artery redox state or if redox conditions regulate NO dependent increases in UBF.
Cystathionine γ-lyase (or cystathionase; CSE) is an E2-sensitive enzyme expressed in uterine arteries that regulates vascular tone and redox state [1,16,17]. CSE is a source of hydrogen sulfide (H2S), a gasotransmitter that induces activating phosphorylation of eNOS, forms vasodilatory NO–H2S hybrid species, and inhibits the sGC-PKG attenuator phosphodiesterase-5 (PDE5) [[18], [19], [20]]. While most uterine artery H2S synthesis is CSE-independent [[21], [22], [23]], CSE is the only mammalian enzyme that produces endogenous cysteine, the limiting substrate for synthesis of the antioxidant glutathione (GSH) [1]. A high GSH abundance and GSH to GSSG (oxidized GSH) ratio are associated with decreased intracellular oxidative stress [24]. Oxidative stress induces endothelial CSE protein accumulation [25], whereas CSE knockout mouse (CSE KO) neurons and hepatocytes produce less GSH and are hypersensitive to H2O2 [16,26]. On the other hand, GSH injection increases femoral artery relaxation by NO in patients with atherosclerosis [27]. Prior work also showed that E2 enhances CSE protein expression in sheep uterine artery myocytes [17], and pregnant CSE KO mice develop hypertension [28]. These findings suggest that uterine artery E2-NO signaling may be sensitive to CSE modulation of redox homeostasis.
In this study, we hypothesized that CSE augments E2-stimulated NO enhancement of UBF by maintaining cysteine and GSH redox homeostasis, which could be important for fertility and pregnancy. We measured blood flow in uterine and renal vessels from ovariectomized wildtype (WT) and CSE KO mice treated with E2 and/or P4. We evaluated NO metabolites and signaling, amino acid substrates, and GSH levels to determine which aspects of E2-stimulated NO signaling might be sensitive to CSE deficiency. We found a critical link between CSE and hormonal regulation of UBF and propose that cysteine-dependent redox homeostasis is a previously unrecognized aspect of uterine artery physiology.
Reagents: Irradiated Global Soy Protein-Free Extrudent mouse chow #2920X was from Envigo-Teklad. E2 (17β-estradiol; #E2758), P4 (#P8783), and all other chemicals were from Millipore Sigma unless stated otherwise.
Animals: The University of Colorado School of Medicine Institutional Animal Care and Use Committee approved all work with mice (Protocol #90). We used 3 to 6-month-old C57/BL6 (WT) and CSE knockout (CSE KO) virgin females housed in a 20 °C vivarium with a 12-hr light/dark cycle and provided ad libitum water and chow, which was 0.3% (w/w) cystine and 0.5% (w/w) methionine. WT mice were C57Bl/6 background from Charles River Laboratories (#027), and CSE KO mutants were backcrossed to the same C57Bl/6 strain for at least 10 generations. CSE KO mice were a kind gift from Prof. Solomon H. Snyder (Johns Hopkins School of Medicine, Baltimore, MD, USA) [29]. All mice were ovariectomized at least 2 weeks prior to hormone treatments. We euthanized mice by CO2 asphyxiation and cervical dislocation.
Uterine and Renal Artery Dopplers: We measured mouse vascular parameters by transcutaneous micro-ultrasound (Visualsonics®) as described before [30]. Briefly, mice were lightly anesthetized with 1–2% (v/v) isoflurane dissolved in 2.5% (v/v) O2, and body temperature was maintained at 37 °C. The sonographer maintained a probe angle of ≤30° to obtain color and pulsed wave spectral Doppler waveforms from the left uterine artery (near the internal iliac artery) or the left renal artery. We determined maximal flow velocity from the average of three consecutive cardiac cycle waveforms. To calculate volumetric blood flow (Q; mL/min), we used the equation Q = (Vm/2)·π·(Dm/2)2, where Vm is maximal velocity (cm/min) and Dm is vessel diameter (cm). This equation assumes a parabolic profile in which mean velocity is half of Vm [31]. Flow data are reported as both absolute (mL/min) and normalized to body weight on the day of measurement (mL/min·kg). To correct for color Doppler artifact [32], we measured vessel diameter in both color and B-mode. Diameter measurements exhibited strong regression (r2 = 0.96) and afforded a reliable correction formula for color image measurements (Supplemental Fig. S1). To estimate technical reproducibility of flow assessment, we calculated coefficients of variation (CV) for vehicle-treated mice on three different days. Repeated measures of flow with color correction exhibited low variability (CV ≤ 13.7%; Supplemental Fig. S1). Heart rates were indistinguishable between genotypes and vessel types for vehicle-treated mice (Table 1). Apart from color Doppler correction and technical control measurements, all other sonography procedures followed the method depicted in Fig. 1.
Hormone treatments: To determine hormone effects on blood flow, we randomized mice to treatment and performed both independent group and paired design analyses. We evaluated uterine artery blood flow of ovariectomized mice at baseline (without treatment) and again after two daily subcutaneous injections of sesame oil vehicle (10 μL/kg), E2 (0.5 mg/kg), P4 (0.5 mg/kg), or both E2 and P4 (Fig. 1A). For independent group analysis, all baseline sonogram measurements were combined. For paired design, only mice with the same before (pre-treatment) and after (two days of exogenous hormone) injections were included.
Tissue and serum collection: Two days after the first vehicle or hormone injection, immediately following the second Doppler ultrasound assessment, mice were euthanized and dissected on ice. We collected serum via left ventricle cardiac puncture, transferred blood to a Serum-Separator-Tube (#365967, BD Biosciences), and centrifuged according to manufacturer's instructions. After removing and weighing the uterus, we excised uterine and renal arteries. Vessels were cleaned of fat and connective tissue under a dissecting scope and flash frozen in liquid N2. Both serum and tissues were stored at −80 °C for future use.
Nitric oxide metabolites (NOx): We quantified serum and uterine artery NOx (total NO2− plus NO3−) using the modified Griess Total Nitric Oxide Assay (#KGE001; R&D Systems) [33]. NOx is a rough proxy for steady state NO synthesis [34].
Uterine artery protein extraction and immunoblotting: Protein was prepared for Western blot as before [35,36]. Briefly, we used steel beads and a Bullet Blender (Next Advance) to homogenize uterine arteries in a lysis buffer containing 50 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA with 2X Protease Inhibitors (#P8340; Millipore Sigma). After centrifuging homogenates at 18,000×g for 10 min, we removed the supernatant and determined protein concentration by BCA assay (#23227; Thermo Fisher). To mitigate low protein yield due to the small size of mouse uterine arteries (5–8 mg/animal), we concentrated 50 μg total lysate protein by precipitating with one volume ice-cold 2x PBS, eight volumes ice-cold acetone, and one volume 6.12 N (100%) trichloroacetic acid. We incubated samples at −20 °C for 1 h, centrifuged at 18,000×g for 10 min, and then washed the protein pellets three times with fresh acetone. After the last centrifugation, protein pellets were air dried at room temperature, suspended in Laemmli buffer (100 mM Tris [pH 8.0], 4% (w/v) SDS, 20% (v/v) glycerol, 200 mM dithiothreitol, 0.05% bromophenol blue), and denatured at 95 °C before SDS-PAGE separation on 4–20% tris-glycine gels. Protein was transferred to Immobilon-FL PVDF membranes (Millipore Sigma), blocked in 2.5% (w/v) milk in PBS, and incubated overnight at 4 °C with the primary antibodies validated previously [35,37]. Primary antibodies detected cyclic GMP-dependent kinase-1 (PKG1; Cell Signaling Technology #3248S, 1:1000), cystathionine β-synthase (CBS; Proteintech #14787-1-AP, 1:2000), cystathionine γ-lyase (CSE; Abnova #H00001491-M01, 1:1000), endothelial NO synthase (eNOS; BD Biosciences #610297, 1:1000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; GeneTex #GTX100118, 1:15,000), phosphodiesterase 5 (PDE5; Cell Signaling Technology #2395S, 1:1000), and soluble guanylate cyclase β1 subunit (sGCβ1; Cayman Chemical #160897, 1:1000). We used IRDye secondary antibodies (Li-Cor #926–32210 and #926–68073; 1:7500). We visualized proteins with an Odyssey CLx (Li-Cor Biosciences) system.
Serum amino acid quantification: We followed methods of de Vrijer et al. [38] with slight modifications to measure serum amino acid concentrations. Serum was collected at euthanasia, two days after the first subcutaneous injection and immediately following the second Doppler ultrasound assessment. We deproteinized serum with 10% sulfosalicylic acid containing 0.3 μmol/mL norleucine as an internal standard. We adjusted pH to 2.2 with 1.5 N LiOH, centrifuged for 3 min at 9100×g, and recovered supernatants. To quantify alanine, arginine, asparagine, aspartate, citrulline, glutamate, glutamine, histidine, isoleucine, leucine, lysine, ornithine, phenylalanine, proline, taurine, threonine, tryptophan, tyrosine, and valine, we analyzed supernatants with a Dionex TM ICS 5000+ high pressure ion chromatograph with a Pickering PCX Pinnacle 120-4 channel variable wavelength detector (Thermo Electron North America, LLC). Amino acid concentration was measured after reaction with ninhydrin (Pickering) for 1 min at 570 nm with Chromeleon chromatographic software (Thermo Electron). To quantify 2-aminobutyrate, cystathionine, total and oxidized cysteine, dimethylglycine, glycine, total homocysteine, methionine, methylglycine, serine, and taurine, we performed capillary stable isotope dilution GC-MS on reduced samples using established methods [39]. The cysteine/cystine ratio was determined by dividing total (reduced) cysteine by oxidized cystine from assays of the same serum samples.
Uterine and renal artery glutathione: We determined uterine and renal artery GSH and GSSG by the method of Jones et al. [40]. Tissue was homogenized by sonication in GSH extraction buffer (5% [w/v] perchloric acid, 0.2 M boric acid, 10 μM γ-Glu-Glu) and pelleted by centrifugation at 18,000×g for 2 min, after which the supernatant was retained and shielded from light. To derivatize sulfhydryl and amino groups, we combined 5 vol of supernatant with 1 vol fresh 40 mM iodoacetic acid, adjusted pH to 9.0 with ~3.3 vol 1.6 M KBH4/KOH, and incubated solutions in the dark for 20 min at room temperature. We then added 5 vol fresh 75 mM dansyl chloride in acetone and incubated solutions in the dark overnight at room temperature. We extracted unreacted dansyl chloride with 9 vol chloroform. The aqueous phase was separated by HPLC using a 3-aminopropyl column with in-line 335 nm excitation and 515 nm fluorescence emission. To quantify GSH and GSSG content, we expressed sample elution peak integrals relative to calibration standards with the same retention times. We normalized GSH and GSSG content to mean values from vehicle-treated WT mice and determined GSH/GSSG ratios using normalized data.
Statistics: We conducted statistical tests with Prism 8.0 (GraphPad; San Diego, CA) with significance set at p < 0.05. We assessed normality with D'Agostino & Pearson tests. We analyzed normally distributed data by Welch's t-tests or one-way ANOVA with Sidak posttests. We analyzed non-normal data by Mann-Whitney tests, Wilcoxon signed rank tests (for paired measures), or Kruskal-Wallis with Dunn posttests. We assessed the relationship between color Doppler and B-mode measurements of arterial diameter by Pearson's correlation and simple linear regression. To calculate coefficients of variation (CV), we divided average weight-normalized blood flow for each mouse by the standard deviation for each mouse then averaged all mice. Supplemental Table 1 data are reported as mean ± standard deviation. All other data are reported as mean ± standard error of the mean (SEM). N refers to the number of individual mice per treatment or assessment.
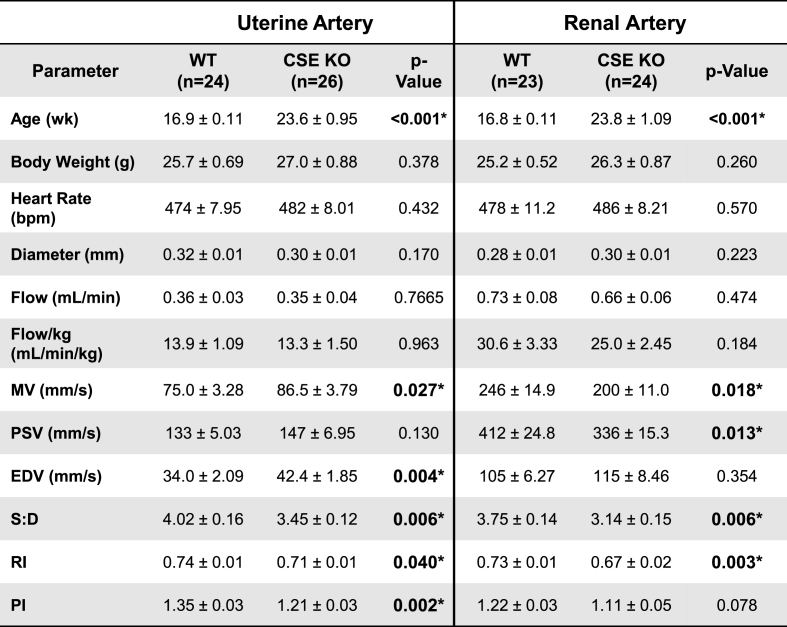
To better understand the role of CSE in uterine physiology, we used color mapping and spectral Doppler ultrasound to measure the velocity, diameter, and calculated blood flow of uterine and renal arteries in ovariectomized WT and CSE KO mice (Fig. 1). There was strong correlation between color and B-mode ultrasound measurements of artery diameter (r2 = 0.96). Calculated blood flow (mL/min*kg) yielded a coefficient of variation of ~10%, demonstrating technical precision (Supplemental Fig. S1) [41]. First, we assessed baseline uterine and renal artery parameters for untreated ovariectomized CSE KO and WT animals. We found no difference between genotypes for heart rate, vessel diameter, or blood flow in either uterine or renal arteries (Table 1). End diastolic velocity in CSE KO uterine arteries was 1.2-fold higher than for WT mice, and CSE KO renal artery peak systolic velocity (PSV) was 1.2-fold lower than for WT mice (Table 1), but total blood flow was not different. These data indicate that small differences between WT and CSE KO vascular indices at baseline (PI, RI; see Fig. 1) are not sufficient to alter uterine or renal artery flow.
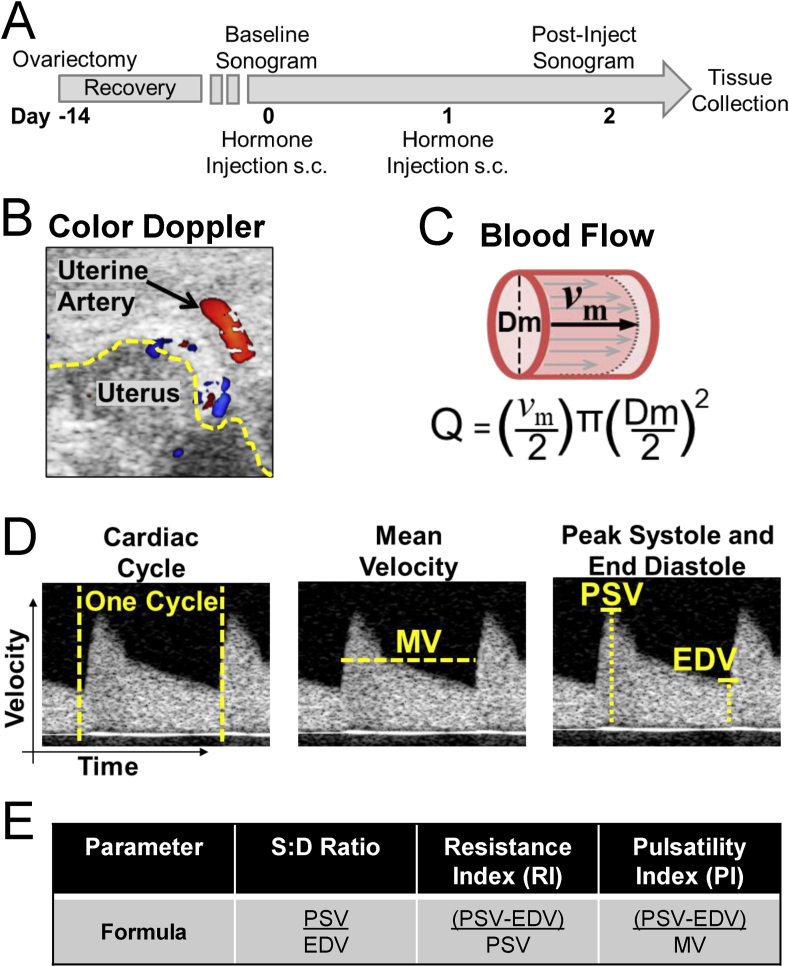
Doppler ultrasound experimental design. A. Experimental design. Ovariectomized WT or CSE KO mice received two daily subcutaneous injections of hormone or sesame oil vehicle. Ultrasounds were conducted just prior to and two days following the first injection, after which mice were euthanized and tissues collected. B. Example color Doppler sonogram of mouse uterine artery. The yellow dashed line denotes uterine horn edge. C. Calculation of volumetric blood flow (Q), which depends on blood vessel diameter (Dm) and average blood velocity. Assuming laminar parabolic flow, average blood velocity is half maximal velocity (vm). D. Uterine artery spectral Doppler scans exemplifying hemodynamic parameters. Cardiac cycle: duration per heartbeat. Mean velocity (MV): average blood speed per cardiac cycle. Peak systolic velocity (PSV) and end diastolic velocity (EDV) are the highest and lowest blood velocities during a cardiac cycle, respectively. E. Derived hemodynamic parameters. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)

Uterine blood flow (UBF) increases with E2 levels and with increased E2 relative to P4 [3]. To assess how CSE influences exogenous hormone stimulation of UBF, we measured UBF by Doppler ultrasound in ovariectomized WT and CSE KO mice before and after treatment with sesame oil (vehicle), E2, P4, or combined E2 and P4 (Fig. 2). Vehicle treatment alone had no effect on UBF for either genotype and did not cause UBF to vary relative to untreated mice (Fig. 2A). As expected, exogenous E2 doubled WT UBF. However, E2 did not significantly affect UBF in CSE KO mice (Fig. 2B). Conversely, P4 decreased WT UBF 2-fold but did not significantly change UBF in the CSE KO (Fig. 2C). Combined E2 and P4 did not significantly affect either genotype, but the pattern of change was similar to E2 treatment alone (Fig. 2D). We obtained similar results using both grouped (Fig. 2) and paired (i.e., before-and-after) analyses (Supplemental Fig. S2). In paired measures analysis, E2 increased WT UBF 1.9-fold while P4 decreased WT UBF 1.5-fold, but neither treatment significantly altered UBF in CSE KO mice. Together, these data suggest that CSE is involved in E2 and P4 hormonal regulation of UBF.
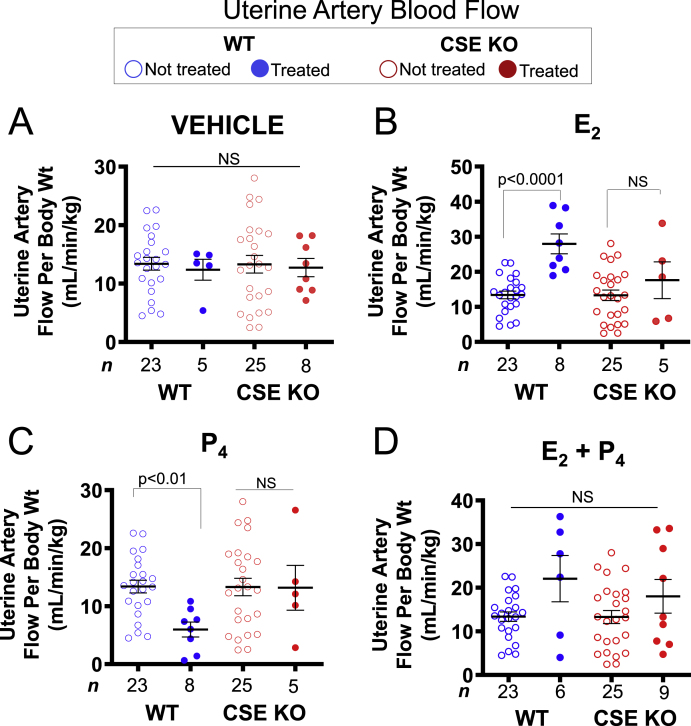
CSE KO uterine artery blood flow is insensitive to both estrogen and progesterone. A. Vehicle treatment (sesame oil) does not affect WT or CSE KO uterine artery blood flow. B. E2 enhances blood flow in WT, but not CSE KO mice. C. P4 attenuates blood flow in WT, but not CSE KO mice. D. Combined E2 and P4 does not affect uterine artery blood flow in WT or CSE KO mice. P-values denote significance by Dunn posttests after non-parametric Kruskal-Wallis tests. NS: not significant. n: number of individual mice (listed in figure). Error bars: SEM.
We wondered whether these uterine artery findings were a global vascular effect of E2 or specific to uterine arteries. Like uterine circulation, renal blood flow can respond to sex hormones. E2 relaxes rat renal arteries contracted with acetylcholine [42]. E2 and P4 also regulate renal catecholamine release, which can change renal blood flow [43]. In contrast to UBF, acute E2 and/or P4 treatment did not influence renal artery blood flow, and there was no difference in the renal artery response to hormones between WT and CSE KO mice (Fig. 3). This finding was evident in both grouped (Fig. 3) and paired analyses (Supplemental Fig. S3). Thus, despite E2 influence on aspects of both uterine and renal physiology, there was a clear selective vascular effect of E2 on UBF. Furthermore, CSE appears dispensable for normal renal artery blood flow and may be uniquely necessary for uterine artery E2 responses.
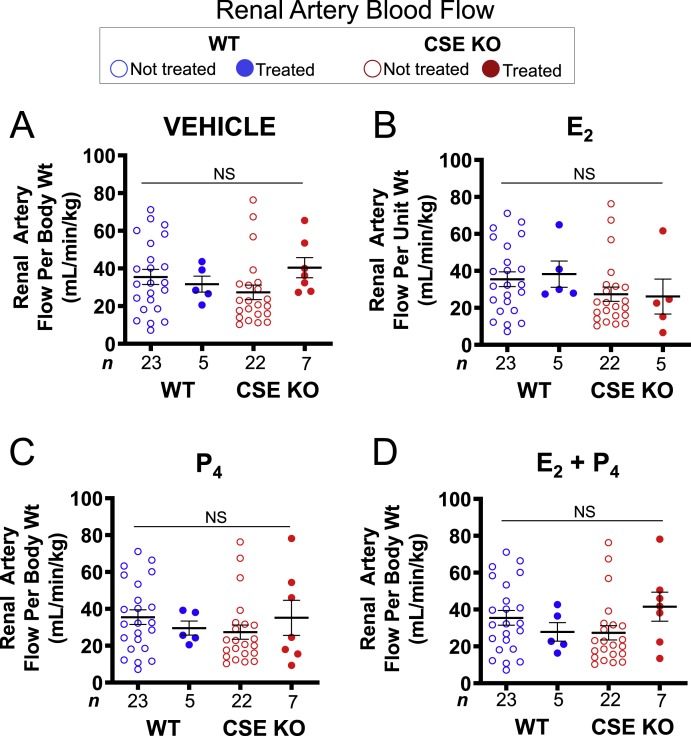
Renal artery blood flow is insensitive to estrogen, progesterone, and the CSE KO mutation. A. Vehicle does not affect WT or CSE KO renal artery blood flow. B. E2 does not affect WT or CSE KO renal artery blood flow. C. P4 does not affect WT or CSE KO renal artery blood flow. D. Combined E2 and P4 does not affect WT or CSE KO renal artery blood flow. NS: not significant by Kruskal-Wallis tests. n: number of individual mice (listed in figure).
E2 and P4 respectively increase and decrease uterine weight [44], and E2 induces myometrial hypertrophy and endometrial hyperplasia [45]. End organ size and metabolic activity are known to increase local blood flow for the mammary gland, skeletal muscle, and kidney [46]. To evaluate whether altered uterine growth alone could explain changes in UBF, we dissected, cleaned, and weighed uteri from WT and CSE KO mice treated with vehicle, E2, P4, or both. Relative to vehicle, WT uterine weight increased 2-fold, decreased 2-fold, and remained the same with E2, P4, and combined treatment, respectively. CSE KO uteri showed similar responses apart from non-significant changes with P4 treatment (Table 2). This indicates that overall uterine growth due to E2 is unaffected by CSE deletion. We also saw no significant differences in uterine weight by genotype within treatment groups (Table 2), indicating that E2 stimulates uterine hypertrophy and hyperplasia similarly in WT and CSE KO animals. This suggests that differences in hormonally regulated UBF are not due to differences in end-organ growth. Thus, distinct mechanisms may regulate E2 effects in the uterus and the uterine artery.
Genotype | Uterine weight (mg) | |||
|---|---|---|---|---|
| Veh | E2 | P4 | E2 + P4 | |
| WT | 37.9 ± 5.9 n = 5 | 75.8 ± 11.8 * n = 8 | 19.4 ± 1.2 * n = 8 | 47.8 ± 3.8 n = 6 |
| CSE KO | 33.0 ± 6.6 n = 8 | 68.9 ± 7.1 * n = 8 | 19.8 ± 1.9 n = 5 | 44.8 ± 4.8 n = 9 |
| p-Value (WT vs. CSE KO) | 0.587 | 0.587 | 0.984 | 0.486 |
NO facilitates E2-dependent uterine artery remodeling and stimulation of UBF [47], and CSE potentiates vascular and myocardial NO signaling [18,19]. Because E2 does not increase UBF in CSE KO mice, we hypothesized that CSE regulates physiologic NO production. To test this, we measured serum total NOx levels (NO3− plus NO2−) from vehicle- and E2-treated WT and CSE KO mice. Unexpectedly, WT serum NOx was E2-unresponsive but E2 decreased CSE KO serum NOx levels 1.7-fold (Fig. 4A). Likewise, WT serum NO3− was E2-insensitive, whereas E2 decreased CSE KO serum NO3− 1.4-fold (Fig. 4B). E2 did not affect serum NO2− levels in either genotype (Fig. 4C), however this NO metabolite has a shorter half-life so that total NOx may be a more stable measure. We then extracted total tissue NOx from dissected cleaned uterine arteries and found a similar relationship. E2 did not affect NOx in WT but decreased NOx 2.5-fold in CSE KO uterine arteries (Fig. 4D). NOx was not significantly different between vehicle-treated WT and CSE KO uterine arteries (Dunn's p-value = 0.07; Fig. 4D). Our findings indicate that E2-stimulated uterine artery NO production or catabolism may be abnormal in CSE KO mice. These data also suggest that uterine artery E2-dependent NO production is influenced by other signaling pathways or metabolites.
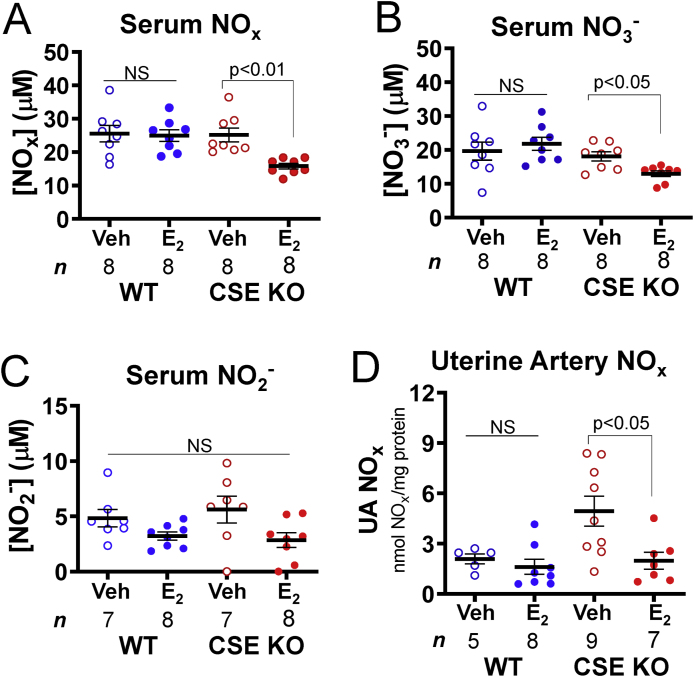
Estrogen decreases serum and uterine artery NO metabolite levels in CSE KO mice. A. E2 injection decreases CSE KO serum NOx levels. B. E2 injection decreases CSE KO serum NO3− levels. C. NO2− levels do not vary with genotype or E2 injection. D. E2 injection decreases CSE KO uterine artery NOx levels. P-values denote significance by Dunn posttests after non-parametric Kruskal-Wallis tests. NS: not significant. n: number of individual mice (listed in figure).
E2 stimulates protein expression and activating phosphorylation of endothelial NO synthase (eNOS), the primary source of vascular NO [47], which stimulates the cGMP-PKG pathway (Fig. 5A). However, uterine artery eNOS protein levels were similar in WT and CSE KO mice regardless of E2 treatment (Fig. 5B and C). Because oxidation state and NO bioavailability can influence downstream signaling and signaling partner expression [35], we also examined the protein levels of other NO signaling partners. Uterine artery sGCβ1, PKG1, and the cGMP signal attenuator PDE5 did not vary with E2 treatment or by genotype (Fig. 5B, D-F). In sheep, E2 enhances uterine artery expression of CSE and cystathionine β-synthase (CBS), another major enzymatic source of H2S [17,22]. We saw no change in the expression of WT uterine artery CSE with E2. Likewise, CBS levels were E2 and genotype insensitive (Fig. 5B, G-H), which may indicate species-specific differences between mice and sheep. Thus, expression of eNOS, NO signaling proteins, CBS, and CSE does not explain the observed functional differences in E2 regulation of uterine artery blood flow.
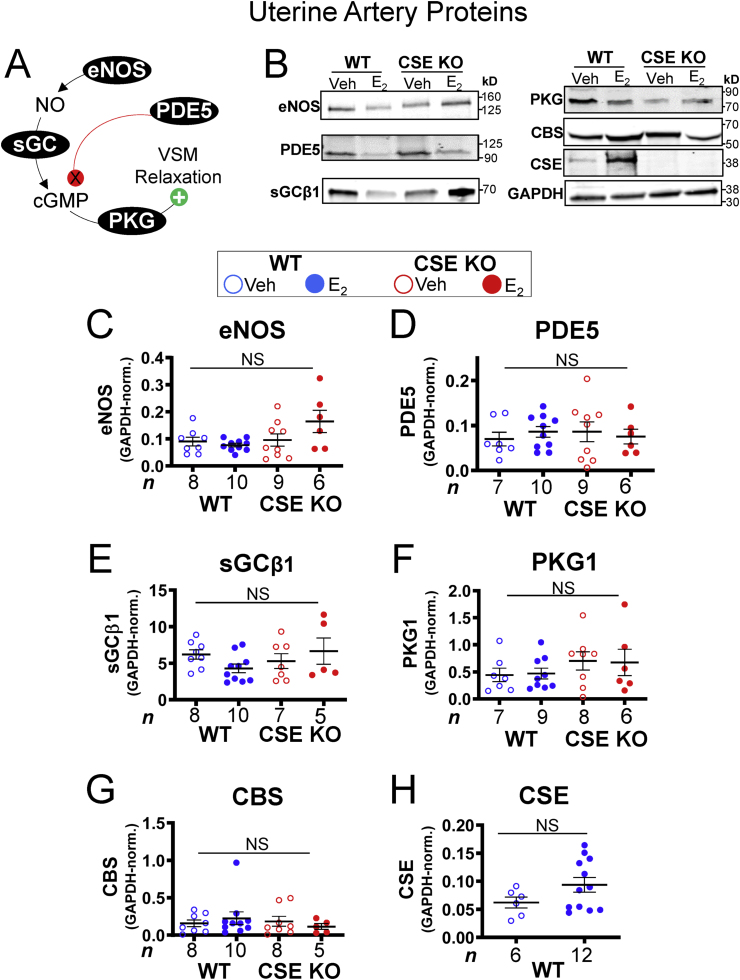
NO-cGMP pathway protein expression is insensitive to estrogen and the CSE KO mutation in uterine arteries. A. Schematic of NO-mediated vascular smooth muscle (VSM) relaxation. NO produced by eNOS activates soluble guanylate cyclase (sGC) to produce cyclic GMP (cGMP), which stimulates protein kinase G (PKG) to phosphorylate multiple targets, thus facilitating VSM relaxation. PDE5 attenuates NO signaling by degrading cGMP. B. E2 does not affect uterine artery protein expression of eNOS, PDE5, sGCβ1, PKG1, or CBS, regardless of genotype. E2 also does not affect CSE protein expression in WT uterine arteries. C–H. Summary data for B. NS: not significant by Kruskal-Wallis tests. n: number of individual mice (listed in figure).
We next assessed whether E2 and CSE regulate serum levels of arginine and its metabolites. NO synthases and arginase metabolize arginine to ornithine and citrulline, respectively (Fig. 6A). Arginine stimulates NO-dependent vasodilation [2], whereas urea cycle arginase competes with NO synthases for arginine, thereby decreasing NOx [48]. Altered abundance of these amino acids could therefore indicate differential urea cycle or eNOS activity. Arginine and ornithine did not vary by genotype or E2 treatment (Fig. 6B and C), suggesting similar substrate availability for NO and urea synthesis in WT and CSE KO serum. E2 did not affect WT or CSE KO citrulline, but citrulline was 2.3-fold more abundant in CSE KO serum regardless of E2 treatment (Fig. 6D). Thus, while E2 does not appear to change overall arginase and eNOS activity, CSE might indirectly increase vascular arginine availability.
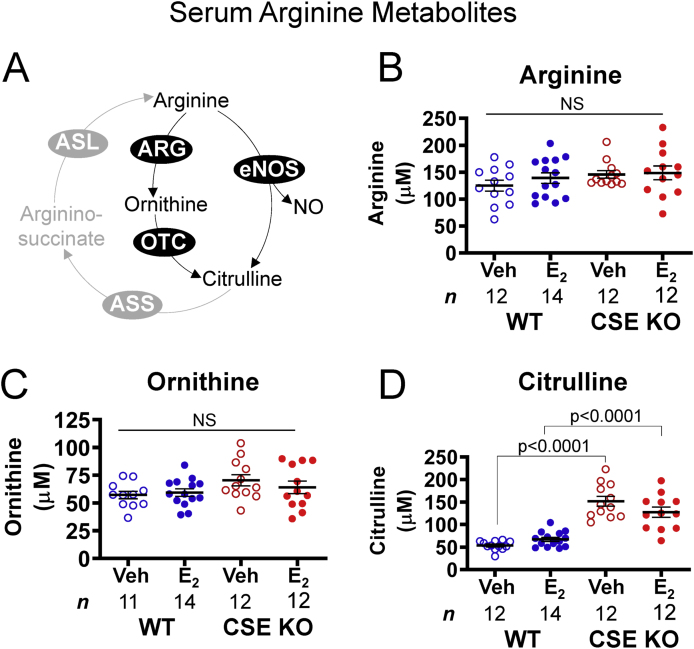
Serum arginine metabolism is disrupted in CSE KO mice. A. Pathways of arginine metabolism. Arginine is a substrate for NO production via endothelial NO synthase (eNOS) and urea elimination via arginase (ARG), which yield citrulline and ornithine, respectively. Ornithine transcarbamylase (OTC) converts ornithine to citrulline. Argininosuccinate synthase (ASS) and argininosuccinate lyase (ASL) regenerate arginine from citrulline. B–C. Serum arginine and ornithine levels do not vary with genotype or E2 injection. D. CSE KO serum contains more citrulline than WT serum, regardless of E2 injection. P-values denote significance by Sidak posttests after one-way ANOVA. NS: not significant. n: number of individual mice (listed in figure).
NO signaling is also sensitive to sulfur amino acid levels. For example, homocysteine inhibits eNOS in endothelial cells [49], whereas taurine antagonizes homocysteine accumulation [50]. As expected from known sulfur amino acid metabolic pathways (Fig. 7A), metabolites upstream of CSE in the transsulfuration pathway were elevated in CSE KO compared with WT serum. Methionine, homocysteine, and cystathionine levels were 1.6-, 26-, and 100-fold higher, respectively, in CSE KO serum (Fig. 7B–D). Conversely, cysteine and taurine levels (downstream of CSE) were respectively lower by 1.4- and 1.25-fold in CSE KO serum (Fig. 7E and F). E2 treatment did not alter sulfur amino acid levels within either genotype (Fig. 7B–F). However, E2 decreased taurine in WT serum sufficiently to eliminate the difference between WT and CSE KO mice (Fig. 7F), similar to our prior report on uterine myometrium [44]. Together, these data demonstrate that CSE deletion increases pro-oxidant homocysteine and decreases antioxidant taurine in serum. Severe hyperhomocysteinemia is associated with diminished NO-dependent vasodilation [49,50], suggesting that CSE could facilitate vascular NO signaling in part by limiting homocysteine accumulation.
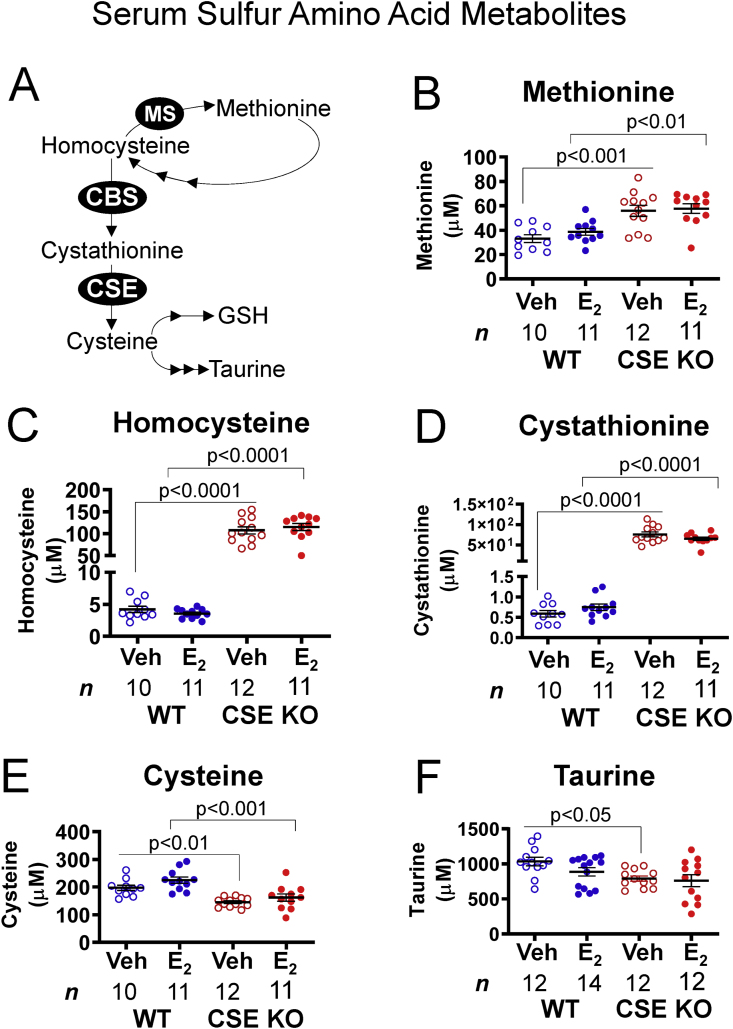
CSE KO mice exhibit altered serum concentrations of sulfur-containing amino acids and associated metabolites. A. Pathways of sulfur amino acid metabolism. Methionine is a precursor for methylation reactions that yield homocysteine. Methionine synthase (MS) regenerates methionine from homocysteine. Alternatively, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) can produce cysteine from homocysteine. Cysteine is a precursor for taurine and glutathione (GSH) biosynthesis. B-D. CSE KO serum contains more methionine, homocysteine, and cystathionine than WT serum, regardless of E2 injection. E. CSE KO serum contains less total cysteine than WT serum, regardless of E2 injection. F. Veh-treated CSE KO serum contains less taurine than veh-treated WT serum. P-values denote significance by Sidak posttests after one-way ANOVA. NS: not significant. n: number of individual mice (listed in figure).
Increased homocysteine and decreased taurine in CSE KO serum is consistent with increased oxidative stress, which is additionally associated with altered abundance of threonine, 2-aminobutyrate (2-AB), and other amino and α-keto acids [51,52]. We therefore measured serum concentrations of 20 amino acids or corresponding α-keto acids from vehicle and E2-treated WT and CSE KO mice. Threonine was 1.3-fold more abundant in CSE KO serum than WT serum with vehicle treatment (p < 0.01); E2 eliminated differences between WT and CSE KO (Supplemental Table S1). Threonine degradation and CSE activity both produce 2-AB, which has been shown to enhance murine myocardial GSH synthesis [52]. While E2 increased 2-AB by 1.4-fold in WT vs. vehicle (p = 0.039), E2 did not alter 2-AB levels in CSE KO serum (Supplemental Table S1). Coincidentally, 2-AB levels were also 1.5-fold lower in CSE KO serum than WT serum with E2 treatment (p < 0.005; Supplemental Table S1). Additionally, the total cysteine/cystine ratio was significantly decreased in E2-treated CSE KO serum (0.41 ± 0.1, N = 3) compared with vehicle-treated WT serum (1.13 ± 0.23, N = 4; Dunn's p < 0.05). This 2.7-fold decrease in the serum cysteine/cystine ratio for CSE KO with E2 treatment may contribute to altered redox homeostasis. Alternatively, it might simply reflect the decreased tissue contribution of endogenously recycled cysteine to the serum pool. In summary, E2 stimulation and CSE deficiency additively perturb serum amino acid abundance in a pattern suggesting altered redox homeostasis.
E2 can inactivate vascular smooth muscle inflammatory NFκB and TNFα signaling and promote flow-mediated vasodilation [3,4,10,11], but E2 stimulation of blood flow can also paradoxically increase vascular oxidative stress through dramatic effects on cellular metabolism [[12], [13], [14]]. Therefore, we tested whether E2 perturbs crucial elements of redox homeostasis in WT and CSE KO uterine and renal arteries. We measured levels of reduced and oxidized glutathione (GSH and GSSG), a critical intracellular antioxidant in eukaryotes [24]. Compared to the WT vehicle condition, neither E2 nor CSE deficiency alone decreased the uterine artery GSH/GSSG ratio, indicating that E2 alone does not diminish redox poise. However, the GSH/GSSG ratio was 2-fold lower in E2-treated CSE KO uterine arteries (Fig. 8A), suggesting CSE deficiency and E2 treatment additively decrease uterine artery GSH redox capacity. Renal artery GSH/GSSG did not vary significantly with any treatments (Fig. 8B). Together these data suggest that E2-induced NO-dependent increases in UBF may require CSE to maintain sufficient intracellular GSH to counteract E2-stimulated oxidative stress in the uterine artery.
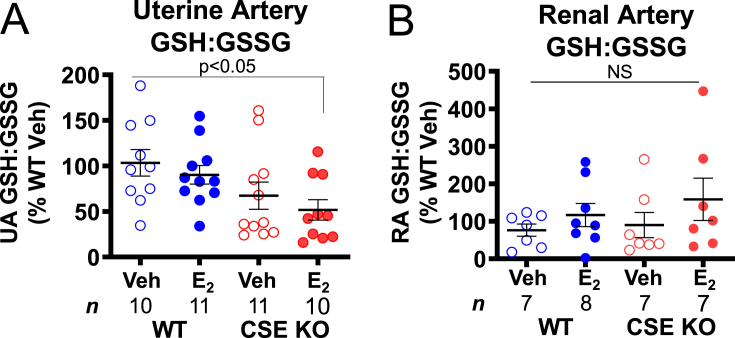
Estrogen decreases the reduced: oxidized glutathione ratio in CSE KO uterine arteries. A. CSE KO uterine arteries contain a lower GSH:GSSG ratio than WT uterine arteries following E2 injection. B. Renal artery GSH:GSSG ratios do not vary by genotype or E2 injection. P-values denote significance by Dunn posttests after non-parametric Kruskal-Wallis tests. NS: not significant. n: number of individual mice (listed in figure).
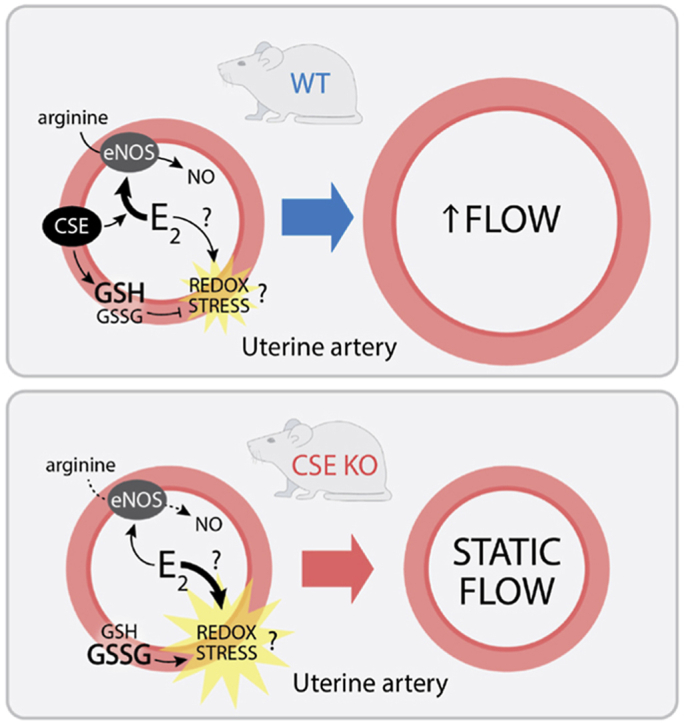
This study used CSE KO mice to identify a critical role for CSE mediating E2 stimulation of UBF. E2 promotes NO-dependent uterine artery vasodilation and remodeling, but also stimulates uterine tissue proliferation that can increase oxidation [3,13,14]. Our data suggest that CSE modulates E2 / NO enhanced UBF by maintaining the GSH/GSSG ratio, which could mitigate E2-induced redox stress (Fig. 9). This novel link between NO and the local uterine artery redox state might inform obstetric vascular conditions such as preeclampsia or fetal growth restriction. Prior literature supports a model for CSE mediating redox homeostasis. CSE deficiency is associated with human placental oxidative stress [53], and neuronal CSE inhibition can deplete GSH and induce cell death [16]. Oxidative stress can also inhibit NO signaling [1]. CSE appears to maintain circulating cysteine levels, increase local uterine artery GSH/GSSG, and prevent loss of uterine artery NOx during exogenous E2 treatment. This could explain why E2 fails to dilate CSE KO uterine arteries and may be a mechanism by which CSE enhances other NO-dependent vessel relaxation [18,19]. We also found that while P4 decreases UBF in WT mice, this does not occur in CSE KO mice. P4 opposes E2 to decrease UBF in non-pregnant cycling cattle [54], and UBF decreases transiently in mice when implantation occurs [55]. While not the focus of this study, it is possible that CSE promotes P4-dependent decreases in UBF during early pregnancy.
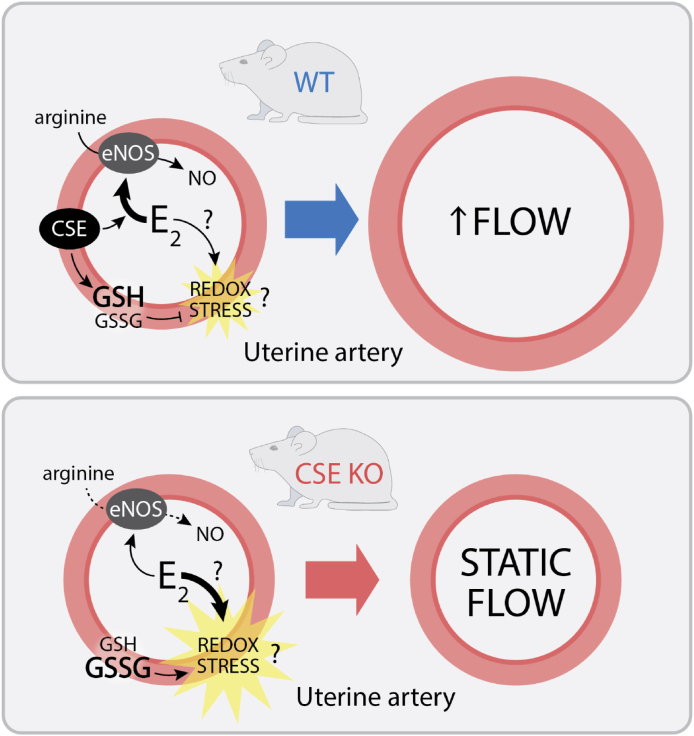
Model for estrogen regulation of CSE-dependent changes in uterine artery blood flow. Upper panel. In wild type (WT) mice, E2 stimulates NO-dependent vascular smooth muscle relaxation, leading to increased uterine artery blood flow. Concomitant with mitogenic activity, E2 also increases oxidative (redox) stress. CSE helps to maintain a high GSH/GSSG ratio, which attenuates oxidative stress in WT mice. Bottom panel. The GSH/GSSG ratio is lower in CSE deficient animals (CSE KO), which leads to greater E2-induced oxidative stress. Oxidative stress attenuates NO-dependent increases in uterine artery blood flow.
CSE regulation of amino acid metabolism may facilitate NO-stimulated UBF. Decreased arginine availability for NO synthesis is a key mechanism of endothelial dysfunction, which may be due to increased endogenous eNOS inhibitors, greater urea cycle activity, or decreased eNOS expression or eNOS S1177 phosphorylation [1,2]. In our model, similar WT and CSE KO serum arginine and ornithine levels suggest comparable urea cycle activity. However, elevated CSE KO citrulline may reflect increased citrulline export from peripheral organs to meet greater endothelial arginine demand similar to macrophages recycling arginine from citrulline for NO synthesis [56]. Consistent with our prior findings in mouse plasma [57], CSE KO mice likely exhibit homocysteinemia due to sex-specific hepatic downregulation of methionine synthase [44,57]. Homocysteine promotes O2− accumulation, which can diminish NO-dependent vasodilation [58]. CBS KO mice are also extremely homocysteinemic [50] which provokes speculation that CBS KO and CSE KO animals might exhibit similar uterine artery dysfunction. However, substantial hepatic injury in CBS KO mice limits their translational relevance for reproductive physiology. Compared with WT animals, CSE KO serum also contains less 2-aminobutyrate (2-AB), a CSE reaction product that pharmacologically promotes myocardial GSH accumulation in mice [52,59]. Elevated homocysteine and diminished 2-AB might therefore contribute to the lower GSH/GSSG in CSE KO uterine arteries. The indistinguishable GSH/GSSG ratios in CSE KO renal arteries could reflect greater total antioxidant capacity in renal arteries than uterine arteries. CSE KO mice have no reproductive phenotype under unstressed conditions [29], consistent with earlier work showing that CSE is especially relevant during episodes of oxidative stress [1,25].
Our findings suggest translational mechanisms by which decreased CSE expression might contribute to vascular obstetric syndromes. Fetal growth restriction and preeclampsia are associated with lower placental CSE protein in humans [60], and blood pressure rises with pharmacologic CSE inhibition in mice [61]. The mechanisms by which CSE promotes vasodilation at the maternal-fetal interface likely involve eNOS. Protein cysteine residues are susceptible to oxidation [62,63], and preeclamptic placentae feature increased eNOS oxidation and decreased NO synthesis [15]. This suggests that uterine artery CSE deficiency could promote preeclampsia via higher oxidative stress and eNOS inactivation. Furthermore, diabetes is a preeclampsia risk factor associated with decreased serum GSH and elevated serum homocysteine [64,65]; homocysteine may increase vascular stiffness in post-menopausal women as well [66]. Hyperglycemia also suppresses CSE protein expression in hepatocytes and peripheral blood mononuclear cells [67]. Given these translational themes, dietary measures to increase circulating l-cysteine might decrease the risk of vascular obstetric syndromes by improving GSH levels to ameliorate oxidative stress in high risk pregnancies. Further work should test these perinatal metabolic hypotheses.
There are several strengths to our study. We used state-of the art techniques such as micro-sonography for vascular Doppler assessment and LC-MS quantification of amino acids. We evaluated different vessel types to compare organ-specific E2 responses in WT and CSE KO mice with the same strain background. We also employed a pharmacologic approach to uncover relationships between UBF and GSH/GSSG in ovariectomized mice using controlled exogenous steroid hormones. Others have observed that E2 promotes uterine artery CSE expression [17] and that CSE KO mice are hypertensive and exhibit increased oxidative stress [26,28]. Our study affirms and extends those findings with a model in which CSE might selectively regulate uterine artery redox state and blood flow. Our study also has several weaknesses. We exclusively used non-pregnant ovariectomized mice, so extrapolation to pregnancy physiology is tentative. CSE regulation of blood flow and vascular redox homeostasis during pregnancy may differ from nonpregnant animals. WT and CSE KO mice, while both C57Bl/6 background, were not sibling-matched, which may have affected within-group differences. In addition, our NOx quantification depended on the gross endpoint Griess assay. NO2− is the first stable reaction product of NO under oxidizing conditions [68], and while serum NO2− levels exceeded the assay's 0.5 μM detection limit [68], uterine artery NO2− was too low to quantify separately. Additionally, we did not assess whether loss of CSE alters eNOS phospho-activation or whether H2S vasodilation differs in CSE KO mice. While CBS produces most uterine artery H2S [[21], [22], [23]], H2S reacts with NO and cGMP to augment NO-dependent vasodilation [1]. Despite these limitations, our study demonstrates that normal sex hormone regulation of NO-stimulated UBF in mice likely requires CSE.
In conclusion, CSE maintains an adequate GSH/GSSG ratio that promotes physiologic E2-NO-dependent uterine artery vasodilation. Our findings in renal arteries suggest that this mechanism is specific to UBF. Differences between WT and CSE KO mouse NOx and amino acid content are also consistent with this model. We plan to determine how relevant environmental stress (e.g., hypoxia) and endogenous E2 and P4 regulate E2 pro- and antioxidant effects in vivo. Likewise, future work with pregnant animals and assessment of vascular histology in WT and CSE KO mice will determine how CSE regulates uterine artery remodeling during pregnancy and whether CSE attenuates uterine artery oxidative damage. Vascular oxidative stress and attenuated vasodilator responses are common to many pathophysiologic processes. Our findings have the potential to improve understanding of disease mechanisms and suggest opportunities for diagnosis and prevention of the Great Obstetrical Vascular Syndromes.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
We are thankful for the University of Colorado Anschutz Medical Campus pre-clinical cardiovascular core for conducting ultrasound measurements with the technical expertise of Maria Cavasin and Tim McKinsey. We also thank Karen Trembler for her assistance completing the amino acid analyses. Ultrasound experiments were supported by NIH grant 1S10ODO18145-01 (Small Animal Ultrasound Imager – Vevo 2100). This work was also supported by a Perinatology-Neonatology T32 training grant (5T32HD007186-37, to DDG), an NIH Shared Instrumentation Grant (S10OD023553, to LB), the William R. Hummel Homocystinuria Research Fund and the Ehst-Hummel-Kaufmann Family Endowed Chair in Inherited Metabolic Disease (to KNM), a
 Cystathionine γ-lyase promotes estrogen-stimulated uterine artery blood flow via glutathione homeostasis
Cystathionine γ-lyase promotes estrogen-stimulated uterine artery blood flow via glutathione homeostasis
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
