
MCP and DF contributed equally to this work.
Despite encouraging progresses achieved in the management of viral diseases, efficient strategies to counteract infections are still required. The current global challenge highlighted the need to develop a rapid and cost-effective strategy to counteract the SARS-CoV-2 pandemic.
Lipid metabolism plays a crucial role in viral infections. Viruses can use the host lipid machinery to support their life cycle and to impair the host immune response. The altered expression of mevalonate pathway-related genes, induced by several viruses, assures survival and spread in host tissue. In some infections, statins, HMG-CoA-reductase inhibitors, reduce cholesterol in the plasma membrane of permissive cells resulting in lower viral titers and failure to internalize the virus. Statins can also counteract viral infections through their immunomodulatory, anti-inflammatory and anti-thrombotic effects. Beyond statins, interfering with the mevalonate pathway could have an adjuvant effect in therapies aimed at mitigating endothelial dysfunction and deregulated inflammation in viral infection.
In this review we depicted the historical and current evidence highlighting how lipid homeostasis and mevalonate pathway targeting represents a valid approach to rapidly neutralize viruses, focusing our attention to their potential use as effective targets to hinder SARS-CoV-2 morbidity and mortality.
Pros and cons of statins and Mevalonate-pathway inhibitors have been also dissected.
Perturbation of lipid homeostasis emerged in the last decade as an important feature during viral infections. As obligatory intracellular pathogens, viruses exploit the host lipid machinery for their entire life cycle, but also alter host lipid metabolism to impair the immune response. Viruses require host lipids for their transport through the membranes, to create a lipid-rich microenvironment for viral genome replication, and use host lipid storage vesicles for their assembly, maturation and egression [1].
The first barrier that viruses run into is the plasma membrane of target cells, which serves as platform to co-ordinate viral entry through different mechanisms. Most viruses interact with membrane lipids that behave directly as receptors or indirectly as cofactors to promote adhesion of the virions, allowing them to explore the cell surface until recognition of specific binding receptors [2]. Various non-enveloped viruses (e.g. polyoma viruses) usually bind to gangliosides, glycosphingolipids with one or more sialic acid residues, to enter the cells. The viral binding to a specific ganglioside induces actin rearrangements that stabilize the virus-ganglioside complex and lead to its inclusion in membrane curvatures [3]. Other viruses (e.g. vesicular stomatitis virus) seem to gain cell entry through interaction with negatively charged phospholipids, like phosphatidylserine [4]. Alternatively, some viruses (e.g. Flaviviridae members) couple with lipoproteins and/or apolipoproteins (ApoE, ApoB) to form lipoviralparticles (LPVs), hybrid structures recognized for internalization by lipid trafficking receptors [5]. The hepatitis C virus (HCV), for example, acquires low-density lipoproteins (LDL) and exploits LDL-receptors (LDL-Rs) for its entry. In addition, the association of LDL with its receptor bring the virus in proximity of other cell-surface receptors with a higher affinity, such as CD81 teraspanin, thus facilitating infection through clathrin-mediated endocytosis. Similarly, LPVs containing high-density lipoproteins (HDL) interact with the scavenger receptor class B type I [6]. For other viruses, such as the influenza A virus (IAV) and the respiratory syncytial virus (RSV), fusion with cell membrane is probably triggered by the presence of certain lipids in the envelope [7]. Following a successful attachment, viruses penetrate the host cell with different mechanisms based on the envelope presence, but the receptor-mediated endocytosis is often the method of choice. The endosomal pathway is strongly dependent on lipids, such as phosphatidylinositol (PI), as signalling molecules to co-ordinate the vesicular events that first bring viruses to the replication site, and then allow the mature virion release. Not surprisingly, the PI3-kinase (PI3K) cascade is activated during virus entry and plays a central role in endosome trafficking and maturation [3]. On another level, lipid composition of endosomal vesicles influences fusion events ending with viral genome delivery [8].
Being essential in membrane fluidity, organization and signalling, cholesterol is certainly one of the most important lipids for viral events occurring at the membrane surface as well as in intracellular processes throughout the infection. Cholesterol is abundant in lipid rafts, plasma membrane domains enriched with surface molecules used by viruses as receptors to invade permissive cells, as mentioned before [3]. Therefore, many viruses including human immunodeficiency virus (HIV), poliovirus, human herpes virus 6 (HHV-6), West Nile virus (WNV) and some members of the Coronaviridae family, rely on lipid rafts integrity for their internalization and, lately, for their assembly[9]. Coronaviruses (CoVs) are enveloped +RNA viruses infecting both animals and humans, particularly relevant because of the current epidemiologic situation. CoVs possess four structural proteins constituting the envelope: the nucleocapsid protein, membrane protein, envelope protein and the S-glycoprotein Spike, that is responsible for the binding of both SARS-CoV and SARS-CoV-2 with angiotensin converting enzyme 2 (ACE2) receptor, residing in cholesterol-rich subdomains on host cell membrane, involved in viral-plasma membrane fusion and endocytosis. Spike protein of SARS-CoV-2 undergoes transmembrane protease serine 2 (TMPRSS2)-mediated cleavage to complete final entry in target cells [10,11]. Thus, it is not surprising that interfering with membrane dynamics may results in failure of CoV internalization [9].
After infection, many positive-strand RNA viruses early recruit cholesterol and other lipids to reorganize host membranes and establish replication structures. In general, viruses from the Togaviridae, Bromoviridae and Nodaviridae families generate spherules, vacuole-like vesicles deriving from small membrane invagination of organelles (endoplasmic reticulum, mitochondria or endolysosomes). Viruses from the Coronaviridae, Arteriviridae and Picornaviridae families generate by protrusion from the endoplasmic reticulum (ER), a network of double-membrane vesicles (DMVs), connected to a complex of convoluted membranes [12]. Despite the great heterogeneity between viruses, lipid homeostasis is modulated by analogous mechanisms to support replication. As a first adaptive strategy, many of them, and in particular +RNA virus such as coronaviruses, take advantage of host lipid metabolism [13] altering the expression of mevalonate-related genes to stimulate de novo sterol synthesis, necessary to ensure not only the membrane reshaping, but also to provide isoprenoid moieties for post-translational modifications of viral proteins. Cholesterol availability within the infected cell is finely regulated also through an increase of its uptake from the extracellular environment and an efficient vesicular transport through cytosolic compartments [1]. In some cases, in addition to the dependence on cholesterol, viral multiplication relies on fatty acids synthesis too. Even more, Dengue virus (DENV) modulates lipid metabolism in a non-canonical way by inducting a form of autophagy that targets lipid droplets, thus promoting the depletion of cellular triglycerides and the release of fatty acids. This results on an increase in β-oxidation and ATP production that stimulate viral replication [14]. Nevertheless, sterol regulatory element-binding proteins (SREBPs) and liver X receptors (LXRs), transcription factors acting as cellular sensors in opposite ways, are the two master regulators of lipid homeostasis. In cytomegalovirus (CMV), HCV and Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infections, the enhanced lipids metabolism is often associated to the increased cleavage of SREBPs precursors into the mature and active forms [15]. SREBPs transcriptional activity results in an up-regulation of lipogenic enzymes, most notably fatty acids synthase (FASN) and the rate-limiting enzyme of the mevalonate pathway, 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) [16]. Moreover, in CMV infection, SREBP1 cleavage is insensitive to high levels of cholesterol, suggesting that the virus is able to circumvent the normal sterol feedback to maintain constitutive lipid amounts during the whole infection [17]. Recently, SREBP2 emerged as a signalling hub for inflammation and cholesterol metabolism in the new pandemic infection by SARS-CoV-2. SREBP2, in fact, has been found to regulate the production of interleukin (IL)-1β and tumor necrosis factor α (TNF-α) cytokines, and to be upregulated in turn by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inflammatory stimulus [18]. It is also reported that SREBP2 is involved in SARS-CoV-2 exocytosis [11]. Of note, cholesterol and fatty acids, as well as cytosolic phospholipase A2α (cPLA2α) and Fatty acid synthase (FASN), are fundamental players in SARS-CoV-2 DMVs formation [11,13].
By contrast, lipid metabolism is found suppressed in CMV-infected macrophages [19] that intensively product an oxidised cholesterol metabolite, the 25-hydroxycholesterol (25-HC), known to have role in inflammation and to significantly attenuate proteolytic processing of SREBP2, thus inhibiting the isoprenoid branch of the mevalonate pathway [20].
Besides cholesterol and fatty acids, distinct phospholipids are required to build functional replicative structures in the late stages of virus life cycles. Some Picornaviridae family members and HCV recruit host phosphatidylinositol 4 (PI4) and the enzymes responsible for its phosphorylation as co-factors at the replication site. Coupling of viral RNA polymerase with PI4-phosphate (PI4P) influence its association with membranes and the ability to synthetize RNA [8].
Overall, the involvement of host lipids in virus life cycle strongly suggests that disrupting lipid homeostasis at multiple levels could help to limit viral invasion and replication. This appears remarkable when more than one strategy is needed to prevent the worsening of viral diseases or to resolve them, just like in the case of epidemic and pandemic coronaviruses.
Several studies showed that both Methyl-β-cyclodextrin (MβCD) and statins, by removing cholesterol from lipid rafts and by reducing cholesterol availability to form membranes, respectively, inhibit early phase of viral infection [[21], [22], [23]]. MβCD and Lovastatin were able to reduce Kaposi's sarcoma associated herpesvirus (KSHV) virion production. Moreover, cholesterol reverted the MβCD-caused reduction of KSHV virions and enhanced the virus release, suggesting a crucial role of lipid rafts in KSHV assembly and egress [24]. In addition, MβCD-mediated cholesterol depletion abolished the binding of gp120 glycoprotein of HIV envelope to CD4/C-C chemokine receptor type 5 (CCR5) receptors located in host’s cells rafts [21] and reduced the binding between Spike protein of SARS-CoV and ACE2 receptor on lipid rafts of the host cells, inhibiting its replication [25]. The use of statins produced positive results also in both enveloped ssRNA Ebola (EBOV) and Zika (ZIKV) viruses. Combination of Atorvastatin and Irbesartan, an Angiotensin receptor blocker (ARB), reduces mortality from pneumonia or sepsis, restoring endothelial cell function and sustaining tissue repair in Ebola patients [26]. It has been demonstrated that statins interfere with viral glycoprotein maturation, limiting EBOV virions infectivity in a human liver cell line and in primary human macrophages [27]. Likewise, in ZIKV-infected Vero cells, lipophilic statins decreased the production of infectious ZIKV particles [28], but the mechanism remains to be clarified.
On the other hand, antiviral effect of statins emerges also in preventing the synthesis of isoprenoid intermediates, required by different viral proteins for their proper anchorage to the rafts [[29], [30], [31]]. In vitro and in vivo studies suggested pleiotropic effects of HMGCR inhibitors against influenza virus (IV), with a mechanism that seems to be strain-dependent. Atorvastatin and Rosuvastatin inhibit Rho/Rho kinase pathway, downregulating H3N2 and H1N1 influenza strains proliferation in Madin Darbyn canine kidney (MDCK) cells. Interesting, Atorvastatin reduces lung viral titer in mouse infected with H3N2 and H1N1 strains [32]. Simvastatin-mediated inhibition of protein prenylation, arrest H1N1 replication altering RhoA and Rab localization, with consequent actin filaments condensation and autophagosomes retardation, respectively [33]. Atorvastatin inhibits IAV replication in vitro, suppressing the production of lipid droplets, thus suggesting that HMGCR inhibitors can play a preventive role in influenza infection, attenuating its severity [34]. Finally, the lipophilic Simvastatin, Atorvastatin and Fluvastatin but not the hydrophilic Pravastatin, have shown a direct effect on HCV RNA replication in vitro, with strong synergism with interferon γ (IFN-γ). This ability seems not related to the lipid lowering effect and HMGCR inhibition, but rather to the inhibition of protein prenylation, indispensable for RNA replication of HCV [35]. Together with inhibition of protein prenylation, statins antagonize Toll-Like Receptor-Myeloid differentiation primary response 88 (TLR-My88) pathway, inhibiting NF-κB pathway activation. The consequent suppression of cytokines and chemokines is mainly associated to their anti-inflammatory effect, also at vascular level [36].
In lung-epithelial carcinoma A549, hematopoietic K562 cells and Peripheral Blood Mononuclear Cells (PBMCs) from health donors, the modulation of endogenous cholesterol synthesis or exogenous cholesterol uptake affect DENV infection. In these models, knock-down of mevalonate (diphospho) decarboxylase (MVD) gene and treatments with statins, Hymeglusin (HMGCoA synthase inhibitor), or Zaragozic acid (Squalene synthetase inhibitor) strongly reduce DENV replication. However, pharmacological inhibition of farnesylation or geranylgeranylation does not affect dengue replication [37], suggesting that inhibition of several enzymes of mevalonate pathway cascade, beyond the statin mediated HMGCR inhibition, could be an interesting approach (Fig. 1A).

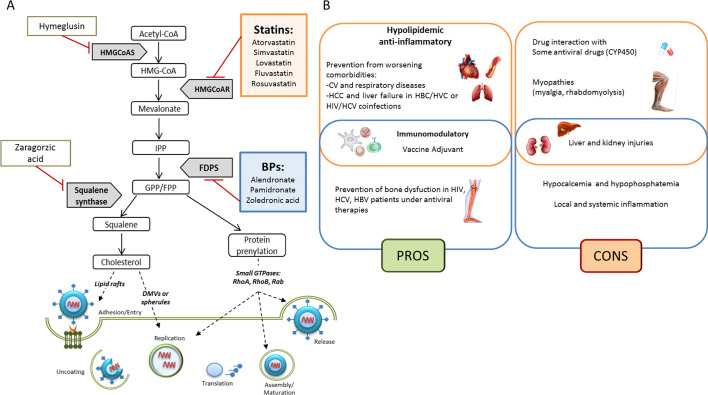
Overview of the mevalonate pathway involvement in viral mechanisms. (A) Pharmacological inhibition of the mevalonate pathway enzymes alters the synthesis of isoprenoid intermediates and of the end-product cholesterol, affecting key steps of viral life cycle. (B) Summary of pros and cons in the use of Statins (orange) and Bisphosphonates (blue) in viral infections. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It has been hypothesized that also bisphosphonates (BPs) such as Zoledronic acid, by inhibiting Farnesyl Diphosphate Synthase (FDPS) and thus the prenylation of small GTPases, could be used to reduce lysosomes release and SARS-CoV-2 virions production. Interestingly, the same author speculated that BPs could be used in Coronavirus Disease 2019 (COVID-19) as immunostimulants and dendritic cells modulators, preventing their ability to activate T-cell [38]. In flu, the efficacy of bisphosphonate Pamidronate, another FDPS inhibitor, has been shown. Through inhibition of FDPS enzymatic activity, Pamidronate reduces lipid raft formation in vitro [39].
Fibrates, synthetic ligands of peroxisome proliferator-activated receptor-α (PPAR-α), used to lower both triglyceride and LDL cholesterol levels, have been shown to have potential antiviral properties [40,41]. Recently, it has been suggested that fenofibrate, by increasing the levels of sulfatide, a glycosphingolipid that negatively regulates virus entry across the cell membrane, could be useful in COVID-19 patients [42]. Moreover, Proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9i) are currently used as lipid-lowering compounds in familial hypercholesterolemic (FH) subjects, to reduce cardiovascular risk. PCSK9i block degradation of LDLR-related protein 1 (LRP1), inducing its availability. Since in CMV infection the increased expression of LRP1 has been associated with reduced infectivity, these compounds have been proposed as eligible drugs to abrogate cardiovascular risk in COVID-19/FH subjects [43]. Yuan and colleagues, by exploring a lipid drug library, showed that AM580, a retinoid acid receptor alpha (RAR-α) agonist, could affect the life cycle of several viruses, including MERS-CoV as well as influenza A virus, through the inhibition of SREBP-related pathways [15].
Sphingolipids and glycosphingolipids are critical molecules not only for membrane integrity but also in immune response, so sphingo-mimetic and sphingo-modulating compounds can be used as therapeutic tool to impact on several aspects of viral infections. Fingolimod (FTY720) is a sphingosine-1-phosphate receptor (S1PR) modulator already used for its immunosuppressive role against multiple sclerosis. The S1PR downregulation produced by Fingolimod, which prevents lymphocytes from exiting the lymphoid tissue, in addition to its antithrombotic and anticoagulant activities [44], have made it a promising drug for the treatment of COVID-19 patients. In fact, Fingolimod is currently undergoing clinical trials for the management of the COVID-19 pandemic (NCT04280588—ClinicalTrials.gov). COVID-19 patients are predisposed to thrombotic events, accompanied by abnormal coagulation and high D-dimer levels. Rosuvastatin and Atorvastatin are able to reduce plasminogen activator inhibitor 1 (PAI1) and tissue factors, strongly involved in the thrombotic complications of Acute Respiratory Distress Syndrome (ARDS), typical of severe COVID disease [36,45]. However, in a randomized, placebo-controlled clinical trial involving patients with sepsis-associated ARDS, Rosuvastatin therapy did not reduce in-hospital mortality neither improve Ventilator- or ICU- free days compared to placebo [46]. Similarly, in a clinical trial including a heterogeneous cohort of patients with non-COVID-19 ARDS, despite safe, Simvastatin did not improve ventilator-free days and other clinical outcomes in ARDS patients [47]. Lately, the identification of two ARDS subphenotypes suggested that the hyperinflammatory ARDS, characterized by increased inflammatory biomarkers and worse clinical outcomes, had a survival benefit from Simvastatin, compared to the hypoinflammatory subphenotype and placebo [48].
In a prospective cohort study in HIV-1-infected adult patients receiving a stable combination of antiretroviral therapy and Rosuvastatin for 12 months, the authors evidenced that the plasma levels of D-dimer, IL-8, and IL-12 are strongly reduced, suggesting a potential role in HIV-induced systemic inflammation [49]. Finally, a recent finding suggests that Simvastatin seems to behave as potent vaccine adjuvant against HA1 influenza, increasing antigen presentation and T-cell activation, in vivo. This effect depends on modulation of post-translational protein prenylation, through HMGCR, FDPS and geranylgeranyl diphosphate synthase inhibition, suggesting that perturbation of mevalonate pathway should be considered also for the improvement of immune response during vaccination [50,51].
Last but not least, in silico studies based on molecular docking, identified statins as potential inhibitors of SARS-CoV-2 main protease (Mpro) [52], thus suggesting that these compounds could potentially affect SARS-CoV-2 life cycle also in a late stage, since Mpro regulates RNA replicase machinery to produce functional viral proteins [53]. In Fig. 1A an overview of mevalonate pathway involvement in viral mechanisms and possible key steps for pharmacological intervention are showed.
The immune system is a complex network of cells and molecules that provides different types of responses to defend the host against pathogens, including bacteria, fungi, and viruses [54]. Viral infections cause the recruitment and activation of both innate and adaptive immune cells that in turn release a wide range of molecules such as cytotoxic granules, pro-inflammatory cytokines, lipid mediators and virus-specific antibodies, that synergistically operate to eradicate the virus [55]. Evolutionarily conserved molecular structures on pathogens, called pathogen-associated molecular patterns (PAMPs), are detected by pattern recognition receptors (PRRs) expressed by the innate cells and trigger intracellular pathways to produce anti-virus immune reactions [55]. For instance, during most viral infections, several PRRs, including TLR7 and TLR9, have been shown to stimulate the production of type I IFNs, a family of pivotal cytokines that induces the transcription of genes involved in host resistance to viral infections [56].
Although innate immune response constitutes the first line of defence against invading viruses, efficient viral clearance from the site of infection and establishment of the protective immunity requires the activation of a virus-specific immune response. In lymphoid tissues, draining sites of infection, virus-specific naïve T lymphocytes are activated by professional antigen presenting cells (APCs) to proliferate and differentiate in effector T (Teff) cells [57]. Upon antigen stimulation, Teff cells can directly destroy virus-infected cells or release pro-inflammatory cytokines, such as, IFN-γ and TNF [58]. In most cases, the immune cell subsets involved are T helper (Th)1 cells, but also Th17 cells may contribute to effector immune responses against viruses, such as HIV, HCV and influenza virus [[59], [60], [61]]. On the contrary, Th2 lymphocytes are rarely associated with inflammatory responses during viral infections, except during severe lung responses RSV [62].
B cells are also increasingly recognized as critical players in the ongoing control of viral infections. Indeed, antibodies produced by B lymphocytes strongly contribute to defend the host, through the binding to infected cells, activation of complement cascade and inflammatory reactions. Further, protective antibodies can be produced even in the complete absence of microbial exposure, thus providing a first line of defence against initial viral infection, defined as natural antibodies [63].
To avoid an excessive inflammatory immune response against viruses, with consequent destructive reactions, immune system has evolved numerous tissue-protective mechanisms that limit the extent of tissue damage [64]. Among these, different regulatory immune cell subsets [[65], [66], [67], [68], [69], [70]], anti-inflammatory cytokines [e.g. IL-10 and tumor growth factor β (TGFβ) [71], chemical mediators (e.g. galectins, resolvins and protectins) [72,73] and inhibitory receptors [e.g. programmed cell death protein 1 (PD1), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), lymphocyte activation gene-3 (LAG3), T cell immunoglobulin and mucin domain- containing protein 3 (TIM3)] [74] have recognized as the main protective mechanisms ensuring the maintenance of immune homeostasis. An important role in maintaining tissue integrity is exerted by CD4+ regulatory T (Treg) cells characterized by the transcription factor forkhead-box P3 (FoxP3), the master gene controlling stability and suppressive functions of this cell subset [75]. Of note, during virus infections more severe inflammatory lesions are observed in mice lacking Treg cells [76]; also, in chronic obstructive pulmonary disease (COPD) subjects characterized by impaired Treg cell frequency and functions [77], respiratory viral infections, especially rhinoviruses, are the major cause of exacerbations [78].
Despite the existence of the above protective mechanisms, some viruses can skip from the balanced immune cell response, causing overt damage to the host. The outcome is influenced by several factors, including biological properties of the infecting virus, type of infection, metabolic and immunological status.
Ongoing research is increasingly focusing on cell metabolism and energetic programs as key intracellular mechanisms needed for immune cell differentiation, growth, and function. It has been reported that specific metabolites, such as glucose, fatty acid and cholesterol provide energy and biosynthetic precursors to support immune responses.
Over the last few years, the impact of cholesterol on the immune cell functions has gained great attention, and cholesterol is now considered a metabolic determinant that may become useful target of novel treatments manipulating the immune response, especially in infectious diseases.
Cholesterol is a critical component in mammalian cell biology, as key molecule of plasma membrane, where it ensures the efficient signal transduction in lipid rafts. Cells obtain cholesterol either by uptake via LDLR, and/or scavenger receptors (e.g., SR-A and CD36), and also through its biosynthesis starting from acetyl-CoA molecules. Indeed, the enzyme HMGCR uses acetyl-CoA and NADPH to generate mevalonate (MVA), the intracellular precursor of cholesterol [79]. As previously mentioned, cellular cholesterol levels are tightly regulated by two transcription factors, LXR and SREBP. LXR acts as nuclear cholesterol sensor activated in response to high levels of intracellular cholesterol, and coordinates cholesterol efflux or cholesterol esterification. On the other hand, low cholesterol levels activate SREBP, which enhances the expression of HMGCR and of the enzymes involved in the cholesterol uptake and synthesis [80].
Studies focusing on cell metabolism have shown that cholesterol pathway has a key role in modulating both innate and adaptive immune cell responses [81,82]. In this context, it is now recognized that the innate immune response depends on cholesterol levels and the activation of Pattern Recognition Receptors (PRRs) and downstream cellular host defense functions are critically sensitive to cellular cholesterol. Experimental evidence reports that inflammatory response of innate cells is regulated by LXR; in macrophages, LXR by promoting cholesterol efflux, represses pro-inflammatory gene expression through effects on NF-κB [83]. However, activation of TLR3 during viral infection inhibits cholesterol efflux from macrophages counteracting anti-inflammatory effects exerted by LXR [84]. In agreement, LXR-null macrophages also display increased susceptibility to pathogen-induced apoptosis [83], and are sensitive to Listeria monocytogenes infection [85].
Recent studies also point on cholesterol pathway as metabolic checkpoints dictating functions of adaptive immune cell subsets [81]. In that regard, a recent study revealed that Epstein-Barr Virus (EBV)-infected B-cells highly engage mevalonate pathway to produce cholesterol and in particular the geranylgeranyl pyrophosphate (GGP), which is important for outgrowth of infected cells [29]. Furthermore, Bensinger et al. found that, in proliferating T lymphocytes, intracellular cholesterol levels are maintained by mutual regulation of the LXR and SREBP transcriptional programs. These authors reported that T cell activation associated with the down-regulation of LXR target genes, involved in cholesterol efflux, and with the simultaneous induction of the SREBP-2 pathway, need for cholesterol synthesis. Of note, activation of LXR prevents mitogen-driven T cell proliferation, while loss of LXR expression confers a proliferative advantage to lymphocytes [86]. Together, these findings suggest that intracellular cholesterol metabolism has a previously unrecognized regulatory role in the control of immune T cell effector functions.
Compelling evidence indicates that SREBP is also important for metabolic reprogramming of CD8+ T cells during viral infection. Indeed, SREBP activity is required to up-regulate glycolysis and oxidative phosphorylation (OXPHOS) metabolism, and to maintain the appropriate intracellular lipid levels to ensure membrane biogenesis and cellular growth [87]. Genetic or pharmacological deletion of SREBP in CD8+ T cells, results in their reduced proliferative ability and impaired expansion during viral infection. Further, the same authors reported that upon infection with Lymphocytic Choriomeningitis Virus (LCMV) SREBP-deficient CD8+ lymphocytes release lower amount of IFN-γ and TNF-α with subsequent inability to counteract viral replication [87].
Piece of evidence reveals that cholesterol metabolism, through the mevalonate pathway, also favours the proliferation and suppressive capability of Treg cells [[88], [89], [90]]. In that regard, Acharya et al. revealed that in vitro supplementation of MVA increases the differentiation of induced Treg (iTreg) cells. Mechanistically, MVA enhances TGF-β signaling by increasing the phosphorylation of small mothers against decapentaplegic (Smad)3, thus promoting FoxP3 expression and suppressive activity [88]. In line with this finding, mevalonate pathway, activated by the serine-threonine kinase LKB1, in response to T cell receptor (TCR) signals, is essential for Treg cell competency and stability; indeed, MVA and its metabolite GGP inhibited the conversion of Treg cells into Th17 cells suppressing the production of IFN-γ and IL-17A [89]. All together, these data confirm the importance of cholesterol and its intermediates in proper function of the immune system, thus interfering with this pathway may represent a potential application to control immune response, particularly during viral infections.
Over the last few years, several pharmacological inhibitors, such as statins and nitrogen-containing BPs have been utilized to control cholesterol metabolism in immune cells. A recent prospective study reported a pro-inflammatory role of rouvastatin in subjects with normal levels of cholesterol [91], despite the large body of evidence that ascribes anti-inflammatory and immunomodulatory properties to statins, suggesting their key role in the treatment of viral infections [92,93]. Among the immunomodulatory roles of statins, it has firstly recognized their ability to repress the major histocompatibility complex (MHC)-II expression on several cell types, such as macrophages, B and activated T lymphocytes [94]. Moreover, it has been shown that statins alter, on T cells, the surface expression of inflammatory molecules, such as lymphocyte function-associated antigen 1 (LFA-1)/intercellular adhesion molecule 1 (ICAM-1) [95] and the CCR5, which are necessary for viral entry [96]. In this context, several studies have shown that lipid rafts are required for HIV-1 infection, thus depletion of cholesterol through statins alters lipid rafts in CD4+ T cell membrane, suppressing viral entry and its replication [97,98]. In agreement, Atorvastatin inhibits HIV-1-infected CD4+ cell division via p21 up-regulation, further preventing virus replication [99].
Statins also control intracellular signal pathway dependent on Rho GTPases [30] and the balance of cytokine production [100]. For instance, it has been demonstrated that statin treatment while affects the induction of Th1 cell response, reducing IFN-γ pathway [101], on the other hand increases the expression of anti-inflammatory Th2 cytokines [102]. Multiple studies indicated that statin treatment in HIV-infected subjects reduces inflammation and activation markers on immune cells, such as C-reactive protein (CRP), soluble CD14, IL-6, IL-8, TNF-α, human leukocyte antigen-DR isotype (HLA-DR) and CD38 [[103], [104], [105], [106]].
Together with direct immunomodulatory activity, it has recently shown that statins contribute to hamper immune cell functions during viral infections. Jameson and colleagues reported that the use of Mevastatin strongly reduced the anti-viral Vgamma9Vdelta2 (Vγ9Vδ2) T cell response against influenza virus, downregulating the production of IFN-γ and the expression of activation markers [107]. Also, the use of Simvastatin, which affects LFA-1 and latent membrane protein 1 (LMP1) lipid rafts [108], inhibits the outgrowth of EBV infected B cells in vitro, further suggesting a possible pharmacological use in human infections [29].
Concerning immune regulatory network, it has been reported that inhibition of mevalonate pathway by statins supports proliferation and suppressive functions of Treg cells during immune activation [90]. In agreement, during HIV-1 infections, Atorvastatin, via mevalonate pathway, expands Treg cells and reduces activation status of Teff cells. Although this finding provides evidence about the immunomodulatory statins activity, possible adverse effects related to their use should be considered. For example, statins attenuate cytotoxic T lymphocytes (CTLs) responses, which are necessary for elimination of HIV-1-infected cells [109].
Even if statins received the greatest attention, other mevalonate pathway inhibitors like BPs have been considered as anti-viral compounds. It has been shown that the FDPS inhibitor Pamidronate expands γδ T cell population and enhances their cytotoxic activity against monocyte-derived macrophages infected with H1N1 influenza virus, thus reducing H1N1 viral titer in vitro and in vivo [110]. Furthermore, it has been shown that in PBMCs from HIV-1-infected subjects, combination of IL-2 and Zoledronate, a bisphosphonate drug, supports γδ T cell activities, and increases DCs maturation and consequent antigen-specific CD8+ T-cell responses [111]. These results suggest the mevalonate pathway blockage as a possible strategy to restore innate and adaptive immune responses.
Additional evidence described that mevalonic acid synthesis is also downregulated by pro-inflammatory IFN cytokines, helping anti-viral immune responses. Indeed, it has been shown that IFNs favors the synthesis of several molecules in macrophages, such as cholesterol-25-hydroxylase (CH25H), viperin, IFN-induced transmembrane protein 3 (IFITIM3), that inhibit virus replication by depleting cholesterol and isoprenoids [112]. Among the IFN-inducible genes, the expression of CH25H was recently correlated to reduced replication of both VSV-SARS-CoV chimeric viruses and a clinical isolate SARS-CoV-2 strain (2019-nCoV/USA-WA1/2020) in vitro [113]. The 25-HC, natural product of cholesterol oxidation by CH25H, modulate cholesterol biosynthesis feedback, act as chemoattractant for adaptive immune cells and as a potent inhibitor for many enveloped virus [114]. Intriguingly, CH25 was found to restrict Spike-mediated fusion at the plasma membrane and the endosomal fusion, thus inhibiting both early stages of SARS-CoV-2 entry into host cells in vitro [113] and viral replication in vivo [115]. Further, several works revealed that IFNs are able to induce the production of specific microRNAs (miR) which can regulate lipoprotein uptake (e.g. miR-125a and miR-455), lipid biosynthetic enzymes (e.g. miR-155, miR-21 and miR-185) and cholesterol efflux (e.g., miR-33 and miR-144) [[116], [117], [118], [119], [120]]. In this context, Robertson et al. observed that the miR-342-5p is induced in macrophages upon treatment with IFN-γ and it regulates sterol biosynthesis by targeting several enzymes involved in the mevalonate pathway, such as SREBF-2, Isopentenyl-Diphosphate Delta Isomerase 1 (IDI1) and Sterol-C4-methyloxidase-like/methylsterol monooxygenase 1 (SC4MOL). Moreover, the authors showed that miR-342-5p in macrophages exerts antiviral properties against multiple pathogenic viruses, including CMV and IAV strain H1N1 [121]. Interestingly, it has been observed that reduction of newly synthesized cholesterol promotes IFNs induction by activating the stimulator of IFN genes (STING), an ER-resident immune receptor and antiviral response against gamma Herpesvirus infection [122]. This finding revealed that cholesterol and IFN pathways communicate as a metabolic-immune circuit, and that ER cholesterol is detected not only by the classical system, but also by innate immune receptors [122]. The effects of cholesterol-lowering drugs on anti-virus immune response are summarized in Fig. 2 .
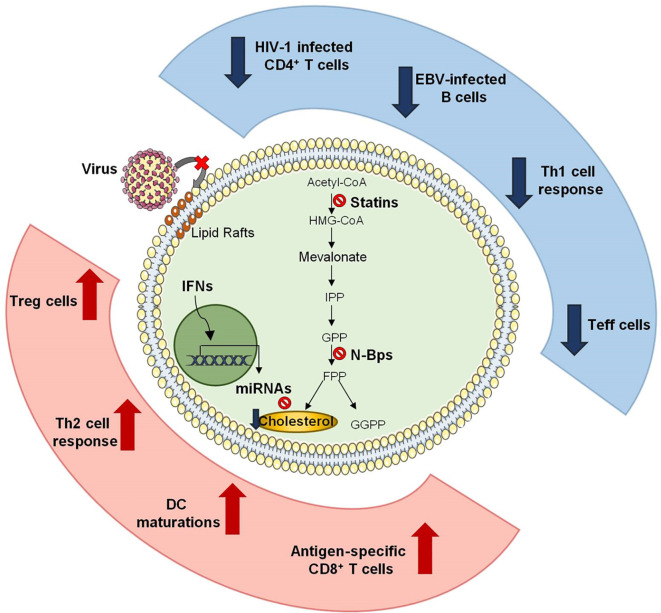
Effects of cholesterol-lowering drugs on anti-virus immune response. Schematic representation of how cholesterol-lowering drugs affect anti-viral immune responses. Depletion of cholesterol by statins alters lipid rafts in CD4+ T lymphocytes and avoids HIV-1 entry and its intracellular replication. Moreover, statin treatment results in the down-regulation of Th1 and in the induction anti-inflammatory Th2 response. During anti-viral immune responses statins also increase frequency and functions of Treg cells while inhibit effector functions of T lymphocytes. Cholesterol inhibition by bisphosphonate determines maturation of antigen presenting cells (APCs) which antigen-specific CD8+ T-cell responses. Finally, IFNs induce the production of specific miRNAs which modulate cholesterol levels in macrophages, thus improving their anti-viral activity.
Despite several evidence highlighted that statins can negatively affect viral processes, some studies suggest that these compounds are ineffective in viruses and their use must be carefully evaluated.
Of note, the cholesterol depletion induced by Lovastatin inhibited hepatitis B surface antigen secretion into culture medium of human hepatoma cells Hep3B, without reducing hepatitis B virions [123]. Simvastatin potentiated anti-HBV activity of Lamivudine, Adefovir, Tenofovir and Entecavir, four licensed anti-HBV compounds. Of note, the addition of MVA did not reduce the efficacy of Lamivudine in combination and was able to revert the anti-HBV effect of Simvastatin [124]. Nevertheless, statins are known to act in the liver by increasing hepatic transaminases in a dose-dependent manner and, although this increase alone is rarely due to liver injury or hepatotoxicity, their use in patients with pre-existing liver disease should deserve caution to avoid worsening of liver function [125,126]. Population-based cohort studies revealed that in patients with HBV infection statins decreased the risk of hepatocellular carcinoma [127] and protected against liver failure during HBV and HCV co-infection [128]. Also, in patients with chronic hepatitis B, Metformin and statins showed a protective effect against hepatocellular carcinoma [129]. A study performed in HIV/HCV co-infected patients, a clinical circumstance strongly associated with a high risk of cirrhosis and hepatocellular carcinoma, concluded that the use of pharmacological HMGCR inhibitors reduces the risk of liver disease progression [130]. Alterations of lipid metabolism and lipogenesis are typical in HCV infections. Noteworthy in HCV-infected mice and humans, hepatic steatosis and hepatocellular carcinoma are ascribable to inhibition of microsomal triglyceride transfer protein (MTP) and activation of SREBP, both processes triggered by the HCV core protein [131]. On the other hand, some authors argued about the possibility to use statins in HCV patients, taking into account their intricate pharmacokinetic and the ability of some HMGCR inhibitors to increase LDL receptor expression, that is essential for HCV entry in hepatocytes [132]. However, in a retrospective study of diabetic or non-diabetic patients with chronic hepatitis C, statin use was associated with improved sustained virologic response (SVR), suggesting a cholesterol-independent mechanism in the antiviral activity of statins against HCV [133]. Furthermore, prospective and retrospective studies highlighted that the combined use of statins with antiviral drugs, such as IFN-α, Ribavirin and protease inhibitors, induced SVR in HCV infection without the occurrence of further adverse effects [134]. Although these clinical data are very encouraging, the use of statins as adjuvants in antiviral therapies should be carefully considered, mainly in HIV and HCV infections. Protease inhibitors and cytochrome P450 3A4 (CYP3A4) inhibitors can alter the metabolism of statins and induce side effects such as myalgia, rhabdomyolysis and renal failure. The lipophilic statins (such as Simvastatin, Atorvastatin and Lovastatin) are mainly metabolised by CYP3A4, Fluvastatin mainly by CYP2C9, while the other statins are mostly excreted unchanged. Furthermore, since both antiviral drugs and statins utilize the same uptake transporter expressed in the hepatocyte membrane OATP1B1, co-administration of these drugs can increase serum concentrations of statins. Therefore, it would be preferable the use of statins that are not significantly metabolized by cytochrome P450. Moreover, in patients taking statins and any other interacting drug, the lipid-lowering effect and the occurrence of adverse muscular events should be monitored [125].
In the light of in vitro findings suggesting that statins prevent caveolar-dependent entry of BK virus (BKV), a Polyomaviridae member, into proximal tubular epithelial cells [23], and clinical studies reporting that statins reduced proteinuria and the progression of kidney disease by improving renal function [135], a retrospective study was conducted in renal transplant recipients with documented BKV viremia taking or not statins to treat hyperlipidaemia. The study failed to demonstrate a beneficial effect of statins, at least at doses maximized for cholesterol lowering, and the authors highlighted the need to perform studies other than Randomized Clinical Trials (RCT) to identify the statins’ doses able to counteract the virus progression [136].
A largely debated point is that statins can be either beneficial against several viruses and, at the same time, they can mediate immunosuppressive effects promoting the reactivation of some latent viral infections. In a case report it has been observed that statins, probably by inducing Treg accumulation, could re-activate HBV infection [137]. Therefore, the increase in Treg can reactivate several latent viral infections. Recently, the use of statins has been associated with increased risk of Varicella zoster virus (VZV) reactivation [138,139], but few and heterogeneous evidence support the link between herpes infections and statin use, without defining a causal relationship [140]. Finally, the immunosuppressive effects induced by statins in haematopoietic cell transplant recipients have been considered a risk factor for human rhinovirus lower respiratory tract infection [141]
Cellular cholesterol is fundamental to support stability and infectivity of several respiratory RNA viruses, including IAV and RSV[142]. Mehrbod and colleagues exhaustively reviewed the effects of mevalonate pathway perturbation in IV infections. Despite some controversies, several clinical studies indicated that statins users are protected against IV infection and symptoms such pneumonia, with relevant mortality rate reduction (40%) [143]. Statin-mediated anti-inflammatory effects lead to decreased production of pro-inflammatory cytokines and reduce the possible development of acute cardiovascular complications in patients with influenza [144]. On the other hand, treatments with different statins in mice infected with highly pathogenic influenza viruses failed to improve overall survival [145]. However, these results could probably depend on the choice of an inadequate dose of viruses for the duration of treatment [143]. Recently, the association between statin use and the clinical outcomes of hospitalized patients with laboratory-confirmed influenza infection has been evaluated. The mortality rates were found similar in statin users and non-users, although statin users were significantly older and had more comorbidities. Furthermore, statin users had fewer complications from the H1N1 influenza virus than non-users, suggesting a protective role for statins in seasonal influenza patients [146]. Of note, lower risks of in-hospital death and hospitalization for pulmonary and circulatory adverse outcomes with influenza vaccination has been observed in statin users, with a slightly lower rate of hospitalization for critical illness. This data suggests that statin use might enhance the protective effects of the vaccine against critical illness [147].
Like statins, also BPs have been investigated as vaccine adjuvants [51]. It seems that statins- or BPs-mediated inhibition of protein prenylation, rather than cholesterol lowering effect, is the mechanism that directly induce innate immune responses [50]. It has been reported that the BPs Clodronate, Etidronate and nitrogen-containing compounds Alendronate and Pamidronate increased both neutralizing IgM and IgG responses against vesicular stomatitis virus (VSV), in infected mice. The adjuvant activity of Clodronate did not depend on γδ T cells or dendritic cells. Moreover, significant increase in total serum IgG levels was detected for up to 3 months upon a single intravenous infusion of BPs, in patients treated for osteoporosis or Paget disease [148].
BPs are used to treat HIV-, HBV- or HCV-related bone dysfunctions such as osteoporosis, to prevent the risk of bone fractures, often induced by anti-viral drugs [149]. A randomized case-control study showed that Alendronate may be able to reverse bone lesions in renal transplant recipients with HCV infection [150] and to increase bone mineral density (BMD) in HIV-infected patients [151]. In a double-blinded, placebo controlled, phase IIB trial, the long-term outcome in Zoledronate-treated patients with HIV infection has been analysed. In patients under antiretroviral therapy, a single intravenous infusion reduced bone resorption and induced BMD starting at 12 and until 144 weeks. No significant changes in adverse effects have been observed in placebo and Zoledronate-treated groups, thus suggesting the safety of Zoledronate as prophylactic antiresorptive therapy in HIV patients [152]. Despite these promising data, the shortage of observational or prospective studies reporting the safety of BPs in other viral diseases, prevents their consideration as anti-viral adjuvant. Improvement of clinical studies is needed to understand the optimal doses, route of administration and the safety especially in co-morbidity-experiencing patients, a clinical condition accounting also for higher incidence of adverse effects. Pros and cons in the use of Statins and BPs in viral infections are summarized in Fig. 1B.
In the last decades Coronaviruses have attracted considerable attention because of the emergencies triggered by the high pathogenic SARS-CoV and MERS-CoV in 2002 and 2012, respectively [153]. While SARS and MERS occurred in limited areas, the new coronavirus SARS-CoV-2 has spread rapidly worldwide causing the current global emergency.
SARS-CoV and MERS-CoV predominantly infect respiratory tract and, unless asymptomatic, produce respiratory illness ranging from mild symptoms, such as fever, to severe progression that culminates in Acute Lung Injury (ALI) and ARDS, often followed by the admission to an Intensive Care Unit (ICU) [154]. It is widely accepted that severe disease is clearly imputable to a series of dysregulation of the host immune response and the so called “cytokine storm”. Indeed, progression to ARDS is associated with high serum levels of pro-inflammatory cytokines (such as IFN-γ, IL-1, IL-6, IL-12, TGFβ) and chemokines (CCL2, CXCL10, CXCL9). In addition, low levels of the anti-inflammatory IL-10 were found [153,154]. However, unlike SARS-CoV and MERS-CoV, high levels of IL-10 and IL-4 seem to be a distinctive characteristic of SARS-CoV2 infection and, in particular, plasma levels of IL-10 were higher in ICU patients compared to non-ICU, suggesting that cytokines profile could define pathogenesis, clinical presentation and natural history of COVID-19 [155].
Several comorbidities influence the course of human diseases caused by CoVs, such as SARS and MERS, as well as COVID-19, the new SARS-CoV-2-associated disease. Beyond the age, the majority of COVID-19 patients underlying diabetes, hypertension, obesity and cardiovascular disease [11,156], suggesting that dysregulations of lipid metabolism and homeostasis play a central role in CoV-diseases.
In the current emergency, many lipid-lowering compounds have been proposed as potential antiviral drugs but, driven by encouraging clinical data, a number of scientists have focused their attention on statins [[157], [158], [159]]. Despite the state of emergency, it is necessary to evaluate the risk/benefit balance of a given therapeutic approach. Taking into account the pleiotropic involvement of mevalonate pathway, its pharmacologic perturbation could potentially produce multiple adverse effects that we need to consider especially, but not exclusively, in frail patients.
Cardiovascular complications seem to have a dual role as both comorbidity and consequence of SARS-CoV-2 infections. Organ distribution of ACE2 receptor (mainly heart, intestine, lung and kidney) strongly influence the course of SARS-CoV-2 infection and it is directly involved in the onset of myocardial injury, myocarditis, cardiomyopathies and coagulopathies [160]. As a matter of fact, after the early phase of the pandemic, coagulopathy emerged as precipitant factor for severe complications and poor prognosis. Furthermore, most COVID-19 patients exhibit endothelial dysfunctions, elevated partial thromboplastin and prothrombin time and general atherothrombotic complications. Additionally, high D-dimer levels and mild thrombocytopenia strongly correlates with the risk of ICU admission and death [158,161]. Hence, there is no doubt that the beneficial effects of statins are ascribable not only to direct inhibition of LDL-mediated inflammation and cytokine storm underlying Cardiovascular diseases (CVDs) and lung injuries, but also to anti-platelet and anti-thrombotic effects that prevent or reduce venous and arterial thrombus formation.
Similarly, to ACE inhibitors (ACEi) and angiotensin receptor blockers (ARBs) currently used in association with lipid-lowering compounds in patients with hypertension and CVDs, statins up-regulate ACE2 expression. However, ACE2 receptor degrades Angiotensin II (AT2), known to possess a pro-inflammatory role, to AT1-7. Once SARS-CoV-2 internalization occurs, ACE2 expression decreases. Consequently, the failed degradation of AT2 causes its accumulation, producing inflammation and tissue damage. Thus, paradoxically, the statins/ACEi/ARBs-mediated ACE2 up-regulation normalizes AT2 levels, resulting in a reduction of inflammation and lung injury [36,158]. At the beginning, many authors supposed that statins-mediated ACE2 upregulation potentially facilitates SARS-CoV-2 entry into cells, conjecturing a worsening effect of COVID-19 in statin users. However, the statin-mediated stabilization of ACE2 receptor seems to contribute to their positive effects in COVID-19.
A series of studies analyzed the association between statins use, prognosis and disease course. In some cases, lipid profile was reported to be altered in close relationship with the outcome of COVID-19 [162]. In a retrospective multicentre cohort study, De Spiegeleer and colleagues concluded that statin users show less COVID-19-related clinical symptoms, compared to non-users, and that the use of statins with ACEi/ARBs is responsible for a better outcome [163]. Another study found a reduced progression to severe disease or death in Atorvastatin users admitted in ICU [164]. Other authors also concluded that chronic statins use correlates with lower in-hospital mortality compared to non-use and they highlighted an overall beneficial and safe effects in COVID-19 patients [[165], [166], [167]]. A large retrospective study including about 14000 subjects, suggested that in-hospital administration of statins significantly reduced the risk of all-cause mortality and the inflammatory response during the hospitalization. Of note, the authors emphasized the safety of the statins – ACEi/ARBs combination in COVID-19 patients [168].
Despite these encouraging studies, data emerging from several meta-analysis revealed conflicting results, certainly due to multiple confounding factors, such as age and cardiovascular comorbidities, influencing data interpretation and preventing adequate conclusions. An early study, including a total of 8990 SARS-CoV-2 patients, confirmed a reduction of about 30% in fatal or severe disease in statin users, compared to non-users [169]. On the other hand, a systematic study including 3449 COVID-19 patients concluded that statins did not improve the outcome of severe disease, but the therapy should be continued in patients with dyslipidaemia since it is safe due to the beneficial effects of statins on cardiovascular complications. However, as the authors acknowledge, the study presents several limits and confounding factors like comorbidities, ongoing therapies, dose and duration of statin therapy [170]. Recently, a meta-analysis including a larger number of SARS-CoV-2 patients confirmed the positive effect of statins in reducing adverse outcome [171]. Of particular interest, Permana and colleagues highlighted that in-hospital use of statins is associated with a reduced mortality risk of about 50% [172]. Importantly, as previously debated, in non-COVID-19 ARDS patients, statins did not improve clinical outcome [46,47], but it seems that ARDS subphenotypes influence the response to statin treatment. In particular, statins seem to be beneficial in hyperinflammatory ARDS [48]. The identification of COVID-19-associated hyperinflammation (COV-HI) subphenotype, characterized by increased C-reactive protein or ferritin concentration and associated with higher mortality, [173] suggests that a proper stratification of COVID-19 patients will be crucial for precision clinical trials design and will help to define and optimize the patient-specific benefits of statin therapy.
Beyond ACE2 upregulation, other issues could limit the use of statins, generically in viral infections and, specifically, in SARS-CoV-2. Reduction of LDL cholesterol levels, inhibition of MyD88 and NF-κB represent an appealing benefit to contrast inflammatory state triggered by viruses. In particular, as previously mentioned, these properties prevent SARS-CoV-2-induced lung injuries and thus severe disease. On the other hand, some authors speculated that statins-mediated reduction of TLR/MyD88 pathway could potentially interfere with the innate immune response, impairing host defence against SARS-CoV-2 [36]. Several authors highlighted that low levels of LDL- and HDL-cholesterol correlate with severe form of COVID-19 [156,174]. This leads to suppose and speculate that statin users should suspend the therapy during hospitalization because it could potentially worsen COVID-19 [175]. However, as debated above, to date no studies have shown worsening of the SARS-CoV-2 disease in statins users, while others demonstrate their safety during the hospitalization [163,164,168].
Noteworthy, myopathies and liver toxicity are among the relevant side effects of statins [176] that we need to consider in COVID-19 patients. Statins-associated muscle symptoms (SAMS) range from myalgia to rhabdomyolysis, with presence or absence of creatine kinase (CK) elevation. Although the pathophysiology of SAMS is not fully understood, mitochondrial impairment has been proposed as a possible cause [177]. Several reports evidenced that statins intake could lower serum and muscle levels of Coenzyme Q10 (CoQ10), also known as ubiquinone 10 in its oxidised form, a lipid product deriving from mevalonate pathway with key roles in mitochondrial respiratory chain and oxidative stress [178]. Therefore, CoQ10 deficiency could possibly lead to mitochondrial dysfunction causing myopathies in statin users [177]. Notably, CoQ10 levels are also decreased in critically ill patients [179] and in metabolic diseases with a clinical picture of inflammation [180], strongly suggesting that CoQ10 levels should be taken into consideration in COVID-19 patients receiving statins. Myalgia, with elevated levels of CK, have been reported to occur in one-quarter to one-half of patients with COVID-19 and it has been proposed as predictive factor for severe outcome [181]. Moreover, in few cases, rhabdomyolysis with high serum levels of CK and liver enzymes, have been observed in severe SARS-CoV-2 infections [[182], [183], [184]]. Although mild liver injury can rarely occur in statin users, liver enzymes levels monitoring is not recommended by FDA, unless statin is combined with other hepatotoxic drugs or liver dysfunction exists [176]. During coronavirus infections, liver damage can occur both because of a direct liver infection or after treatment with hepatotoxic drugs. In COVID-19 patients, serum levels increase of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) enzymes correlates with severity and disease progression, with an increase higher than 15 times the upper normal limit value [185]. Hence, despite they seem to be safe, it would be advisable to discontinue statin therapy in COVID-19 patients with myopathies and liver dysfunctions. Moreover, interruption of statin therapy should be considered also in the case of concomitant treatment of COVID-19 patients with some antiviral drugs, such as the protease inhibitors Lopinavir and Ritonavir. Indeed, the combination of statins with Lopinavir or Ritonavir could potentially inhibit statins metabolism, increasing their serum levels and triggering in turn liver and renal injuries that predispose to worse prognosis in SARS-CoV-2 infection. It has been proposed that, to bypass toxicity, antiviral drugs could be used in combination with Atorvastatin and Rosuvastatin at maximum dose of 20 mg/day and 10 mg/day, respectively [36]. By contrast, some authors highlighted that statins withdrawal often exacerbates the prognosis of SARS-CoV-2 infection. Antithrombotic and anti-inflammatory effects of statins are favourable in COVID-19 patients, since alterations in the coagulation system might occur after SARS-CoV-2 infection. It has been observed that in-hospital interruption of statin therapy leads to rebound effects that amplify cytokine levels and cardiac risk, with the onset of acute coronary deleterious events. Moreover, it seems that statin withdrawal after hospital admission increases mortality of COVID-19 patients [186,187]. On balance, statin discontinuation in COVID-19 patients should be considered with extreme caution.
Like statins, other inhibitors of mevalonate pathway should be considered to manage SARS-CoV-2 infection. In a mouse model of SARS-CoV, Clodronate can deplete alveolar macrophage and directly activate respiratory dendritic cells (DC), triggering the immune response and improving the anti-virus T cell response [188].
In the previous sections we already debated that Zoledronic acid (ZA) could potentially have protective and prognosis-improving properties in COVID-19, acting as immunostimulant able to expand γδ T-cells and to increase activity of dendritic cells. Moreover, ZA-mediated inhibition of protein prenylation leads to speculate that it can be used as inhibitor of SARS-CoV-2 virions release [38]. The hypothesis of Brufsky and colleagues raises from the observation that in severe COVID-19 patients γδ T-cells, DC and NK are depleted [189]. Thus, treatment with compounds able to positively modulate immunopathology of SARS-CoV-2 infection, should be seriously considered. However, clinical data are urgently needed.
As synthetic analogues of pyrophosphate, BPs like Pamidronate, Clodronate and ZA are widely used for the treatment of osteoporosis, hypercalcemia, Paget’s disease, osteolytic bone metastases from breast cancer, and osteolytic lesions from multiple myeloma [190]. Hyperinflammation in COVID-19 patients play a relevant role also in bone physiology. Some cytokines and chemokine like CXCL10, TNF-α, IL-1β and IL-6, secreted during infection, decrease osteoblast proliferation and differentiation, leading to a clear reduction in BMD. In SARS-CoV-2 infected patients, osteonecrosis and reduced BMD has been found and linked to the treatment with corticosteroids during the initial infection. However, reduced BMD has been found also in severe SARS, independently of corticosteroids [181], suggesting that bone disorders should not be overlooked.
The evidence described above, work in favour of BP use in COVID-19 patients, also as vaccine adjuvant. However, several unfavourable issues in the use of BPs need to be considered. First, several adverse effects of BPs could certainly be detrimental in severe SARS-CoV-2 patients, potentially worsening their prognosis. Of note, local and systemic inflammatory reactions have been observed in patients treated with several BPs, accompanied with fever, pain, myalgia, arthralgia and edema, mainly after intravenous administration. In rare cases, acute dyspnea, pneumonitis and pulmonary edema have been observed, as result of systemic inflammation. ZA and Pamidronate have been associated with acute and chronic renal failure or nephrotic syndrome. In addition, BPs can induce electrolyte abnormalities like hypocalcemia and hypophosphatemia [191]. Due to similar mechanisms controlling vascular calcification and bone mineralization and to their lipid-lowering action, BPs have been proposed as inhibitors of atherosclerosis and vascular calcification. Many controversies exist about the efficacy of BPs in reducing cardiovascular complications, but a meta-analysis revealed that, except for ZA displaying a modest increase in the risk of atrial fibrillation, the use of BPs is not discouraged since they did not show significant changes in cardiovascular disease risk [192]. On the other hand, during the current emergency treatment of osteoporosis has not been discontinued, indeed, several guidelines have been proposed for patients under treatment [193]. Finally, the administration of BPs, mainly oral tablets, requires particular conditions that could not favour patient compliance, in particular in ICU admitted COVID patients.
Overall, our critical analysis highlights the beneficial effects of statins and mevalonate pathway inhibitors (MVAi) in COVID-19 patients (Fig. 3 ). Almost all the data demonstrate that statins protect and prevent severe outcome in SARS-CoV-2 infected statin-users. What remains to be clarified is the risk/benefit balance for statin-naïve patients during infection, and the debatable potential role of statins in the prophylaxis. To this aim, well-designed Randomized Clinical Trials (RCTs) are urgently required, also to establish the optimal therapeutic protocol for statin use, in terms of dose, time and type, comorbidities and disease stage.
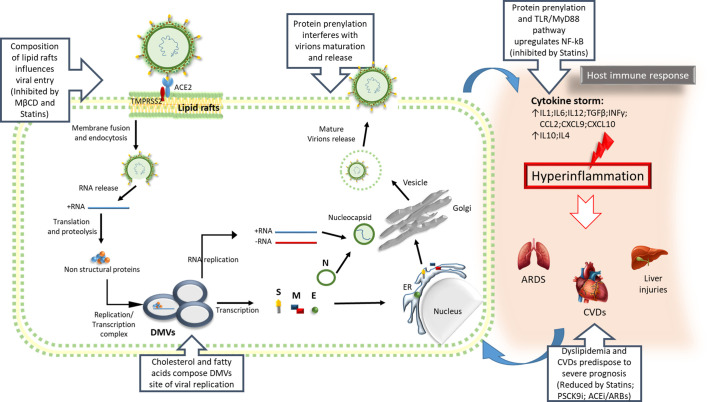
Critical steps of SARS-CoV-2 infection involving lipid homeostasis. Some key steps of SARS-CoV-2 infection, ranging from the virus life cycle to the inflammatory response, can be affected by targeting the mevalonate pathway, making it a potential therapeutic target to counteract COVID-19.
S, Spike protein; M, Membrane proteins; E, Envelope protein; N, Nucleocapsid protein; ER, Endoplasmic reticulum; DMVs, Double Membrane vesicles; CVDs, Cardiovascular diseases.
Despite BPs appear to be promising, observational and prospective studies will help to analyse and consider their potential in COVID-19 disease.
An interesting observation derives from the possibility to use MVAi as vaccine booster also in SARS-CoV-2 infection. Of course, this application must be supported by solid pre-clinical and clinical evidence to consider them as future anti-SARS-CoV-2 vaccine adjuvant.
Development of vaccines against SARS-CoV-2, gives us hope for a definitive and long-term solution, but the current global emergency taught that we need to be prepared for a prompt response to emergencies.
Due to its large involvement in several cellular processes, it is clear that lipid homeostasis represents a sort of Achilles heel for the hosts during the infections by many obligate parasites, including viruses. All the clinical studies from SARS, MERS and SARS-CoV-2 patients highlighted that a clear alteration in lipids assets occurs during infection. This is not surprising since it is strictly linked to almost all the comorbidities predisposing to worst prognosis, such as cardiovascular diseases, obesity, hypertension and others. Then, lipid profile cannot be overlooked for deciphering COVID-19 disease.
Several lipid-lowering compounds were considered for COVID-19 treatment, but statins received the greatest attention because they are low-cost and safety drugs, widely used worldwide. In the last decades a number of studies highlighted the statins pleiotropic effects, also in viral infections. In the last months some, but still few observational and interventional clinical trials evaluated the association between lipid alterations, statin use and disease severity/progression (e.g. NCT04380402; NCT04348695; NCT04333407; NCT04407273; NCT04426084). However, most of these studies have limits related to the high number of unavoidable bias and confounders.
In conclusion, it would be reasonable to extend and deepen well designed clinical studies to decipher the complex metabolic and pathophysiologic processes of SARS-CoV-2 disease, paying more attention to lipid homeostasis dysfunctions. Moreover, in our opinion, pre-clinical studies could also help to optimize the use of statins or other lipid-lowering drugs in these patients, providing valuable guidance on patient-specific use, based on metabolic-associated preconditions.
This study was partially supported by
The authors declare no conflicts of interest.
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
 Lipid homeostasis and mevalonate pathway in COVID-19: Basic concepts and potential therapeutic targets
Lipid homeostasis and mevalonate pathway in COVID-19: Basic concepts and potential therapeutic targets
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp
