
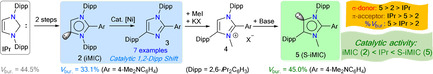
Intramolecular 1,2‐Dipp migration of seven mesoionic carbenes (iMICAr) 2 a–g (iMICAr=ArC{N(Dipp)}2CHC; Ar=aryl; Dipp=2,6‐iPr2C6H3) under nickel catalysis to give 1,3‐imidazoles (IMDAr) 3 a–g (IMDAr=ArC{N(Dipp)CHC(Dipp)N}) has been reported. The formation of 3 indicates the cleavage of an N−CDipp bond and the subsequent formation of a C−CDipp bond in 2, which is unprecedented in NHC chemistry. The use of 3 in accessing super‐iMICs (5) (S‐iMIC=ArC{N(Dipp)N(Me)C(Dipp)}C) has been shown with selenium (6), gold (7), and palladium (8) compounds. The quantification of the stereoelectronic properties reveals the superior σ‐donor strength of 5 compared to that of classical NHCs. Remarkably, the percentage buried volume of 5 (%Vbur=45) is the largest known amongst thus far reported iMICs. Catalytic studies show a remarkable activity of 5, which is consistent with their auspicious stereoelectronic features.
Super‐iMICs (5) with the largest buried volume (V bur=45 %) known thus far for iMICs are accessible via unprecedented Ni‐catalyzed intramolecular 1,2‐aryl migration of C5‐protonated iMICs (2) to 3, subsequent N‐alkylation of 3 to 4, and the deprotonation of 4. S‐iMICs (5) are stronger σ‐donors and superior π‐acceptors than 2. A comparative catalytic study reveals superior activity of S‐iMICs (5) over iMICs (2) and NHC (IPr).
N‐Heterocyclic carbenes (NHCs) (I, Figure 1) are most versatile carbon‐donor neutral ligands in synthesis and catalysis [1] as well as in materials science. [2] This is largely attributed to the auspicious stereoelectronic properties of NHCs. [3] A clear insight into the stereoelectronic properties of NHCs[ 3 , 4 ] facilitates the rational choice of an NHC for a particular application. [5] Mesoionic carbenes (iMICs) (II, “i” refers to 1,3‐imidazole‐derived) are a subclass of the family of NHCs with the carbenic carbon atom at the unusual C4 (or C5) position. [6] iMICs (II) (also known as abnormal NHCs) [7] are stronger σ‐donors than classical NHCs (I), and hence have a significant potential in synthesis and catalysis.[ 6 , 7 ] However, rational synthetic methods for iMIC‐compounds remain extremely scarce compared to those of NHCs. [7a] In 2001, Crabtree et al. reported the first iMIC complex (III). [8] The first crystalline NHC was isolated by Arduengo et al. in 1991, [9] but it was only in 2009 when Bertrand et al. prepared the first unmasked iMIC (IV). [10]

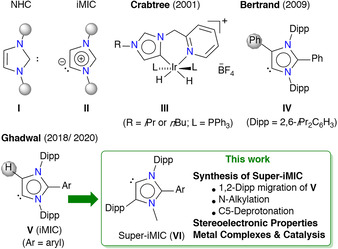
Classical NHCs (I) and iMICs (II). First examples of structurally characterized iMIC–metal complex (III), free iMIC (IV), and C5‐protonated iMIC (V). This work: S‐iMIC (VI).
Like IV, the use of an aryl substituent at the C2‐position is shown to be the reliable strategy for accessing iMIC‐compounds.[ 6 , 7 ] We previously reported two catalytic protocols for the direct C2‐arylation of NHCs to C2‐arylated 1,3‐imidazoli(ni)um salts [11] and revealed their suitability in preparing C5‐protonated free iMICs (V), [12] metal complexes, [13] and stable radicals. [14] In addition to the σ‐donor properties, the steric profile of ligands also plays a key role in the stability and catalytic activity of derived complexes. [1] However, experimental tools to improve the properties of iMICs remained virtually unknown. [7] Herein, we report an unprecedented 1,2‐Dipp migration in C5‐protonated iMICs (V) under nickel catalysis to afford 1,2,4‐triaryl‐1,3‐imidazoles (IMDAr) and reveal their utility in accessing super bulky S‐iMICs (VI) (Figure 1). Besides the quantification of the stereoelectronic properties, the catalytic activity of VI has been probed and compared with those of V and IPr (IPr=1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene).
The deprotonation of C2‐arylated 1,3‐imidazolium salts (1 a–g)Br affords the desired iMICs 2 a–g (Scheme 1 a). [12] Treatment of a toluene solution of 2 a with 1 equiv. of Ni(cod)2 (cod=1,5‐cyclooctadiene) led to the formation of an unexpected product 3 a in 35 % yield (Scheme 1 a), which indicates the migration of a Dipp substituent of 2 a to the carbenic carbon atom to give 3 a. This type of reactivity is unprecedented in NHC chemistry. [15] The exact mechanism for the formation of 3 a is currently unknown. The stronger σ‐donor property of iMICPh (2 a) [4c] probably makes the Ni0 center in the putative complex A electron‐rich (Scheme 1 b). [16] Oxidative addition of Ni0 into the adjacent C−N bond via B is likely to form a NiII species C, which eventually undergoes reductive elimination to yield 3 a. Note, (iMICAr)Ni(CO)3 complexes comprising π‐acidic CO ligands are stable solids. [4c] The stoichiometric conversion of 2 a into 3 a can be translated into a catalytic process that is also applicable to other iMICs (2 b–g). Treatment of a freshly prepared toluene solution of iMICs (2 a–g) with 5 mol % of Ni(cod)2 yields the anticipated compounds 3 a–g in 54–97 % yield (Scheme 1 c). Interestingly, no conversion of 2 a into 3 a was observed in THF (Table 1, entries 1 and 2), instead an intractable mixture of products was formed. In addition to nBuLi, KN(SiMe3)2 can also be used as a base (Table 1, entry 4). Addition of a small amount of cod (10 mol %) to the reaction mixture improves the yield (Table 1, entries 6 and 7). 3 a–g have been characterized by NMR spectroscopy and mass spectrometry. Solid‐state molecular structures of some selected compounds have been determined by X‐ray diffraction (see the Supporting Information). Consistent with the molecular structure (Scheme 1 c), the 1H NMR spectrum of 3 a exhibits two septets and four doublets for the CHMe2 groups. The backbone proton of 3 a (6.82 ppm) resonates at a higher field compared to that of (1 a)Br (8.38 ppm). [11]
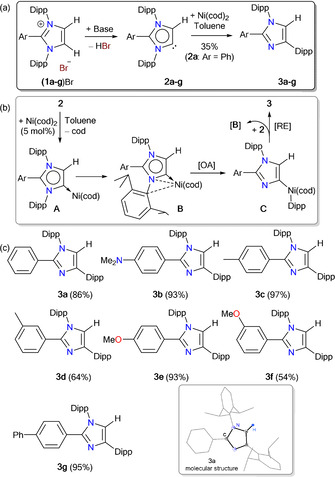
a) Catalytic conversion of 2 a–g into 3 a–g. b) Mechanistic proposal for 1,2‐Dipp shift of 2. c) Isolated examples of 3. Inset: Solid‐state molecular structure of 3 a (thermal ellipsoids at 50 % probability).

|
Entry |
(1 a)Br [g] |
Solvent |
Base |
t [h] |
Yield[b] [%] |
|---|---|---|---|---|---|
|
1 |
0.5 |
THF |
nBuLi |
19 |
– |
|
2 |
0.5 |
THF |
KN(SiMe3)2 |
19 |
– |
|
3 |
1.0 |
Tol |
nBuLi |
19 |
54 |
|
4 |
1.0 |
Tol |
KN(SiMe3)2 |
21 |
93 |
|
5 |
2.5 |
Tol |
nBuLi |
20 |
63 |
|
6[c] |
1.0 |
Tol |
nBuLi |
17 |
92 |
|
7[c] |
2.5 |
Tol |
nBuLi |
17 |
86 |
[a] All reactions to prepare 2 a were performed at −40 °C and after 30 minutes stirring at room temperature Ni(cod)2 was added at −20 °C. [b] Yields refer to isolated compounds. [c] With 10 mol % of 1,5‐cyclooctadiene (cod) as an additive.
Compounds 3 a–g featuring bulky Dipp substituents at the 1,4‐positions are promising starting materials of new sterically demanding iMICs. The direct N‐methylation of 3 a–g affords 1,3‐imidazolium salts (4 a–g)X (X=PF6, I, OTf, or BF4) in good to excellent yields (Scheme 2). (4 a–f)X were characterized by NMR spectroscopy, mass spectrometry, and X‐ray diffraction studies (see the Supporting Information). Selenium compounds have proven appropriate candidates to analyze the electronic properties of singlet carbenes.[ 3 , 4 , 17 ] 6 a and 6 b were prepared by the treatment of (4 a)PF6 and (4 b)PF6 with Se powder and KN(SiMe3)2, respectively (Scheme 2). Compound 7 was isolated as a white solid using a freshly prepared toluene solution of 5 b and (Me2S)AuCl. Similarly, 8‐H and 8‐Ph were obtained by treating 5 b with 0.5 equiv of [(allyl)PdCl]2 and [(cin)PdCl]2, respectively. The 77Se{1H} NMR spectra of 6 a (+9.0 ppm) and 6 b (+2.0 ppm) exhibit a lower‐field signal compared to that of (2 a)Se (−3.0 ppm) and (2 b)Se (−15.1 ppm) containing a C5‐protonated iMIC (2 a,b) (Table 2). [4c] The C2–Se1 bond length and N1‐C2‐C3 bond angle of 6 a (1.856(2) Å, 104.8(1)°) (Figure 2) are comparable with that of(2 a)Se (1.859(2) Å, 104.3(1)°). [4c] The C2–Au1 bond lengths and the N1‐C2‐C3 bond angles of 7 (1.983(2) Å/104.3(1)°) and (2 g)AuCl (2.008(4) Å)/(102.5(1)°) [12a] are fully consistent. The C2–Pd1 bond length of 8‐H (2.042(2) Å) is slightly larger compared to those of other iMIC‐Pd complexes (1.955(2)–2.030(2) Å), [13c] which may be due to steric reason. [18]
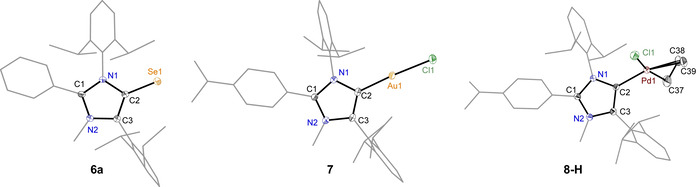
Solid state molecular structures of 6 a, 7, and 8‐H (thermal ellipsoids at 50 % probability).
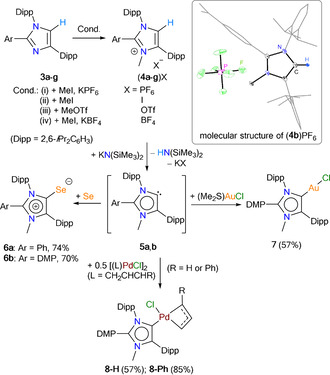
Synthesis of (4 a–f)X, 6 a, 6 b, 7, and 8‐R (R=H or Ph). Only 5 a,b were used to prepare 6–8. Inset: Solid‐state molecular structure of (4 b)PF6 (thermal ellipsoids at 50 % probability).
|
|
77Se[a] [ppm] |
J C–Se [a] [Hz] |
%V bur |
HOMO [eV][b] |
LUMO [eV][b] |
1st PA[c] [kcal mol−1] |
|---|---|---|---|---|---|---|
|
2 a |
−3.0 |
214 |
32.6[d] |
−5.065 |
−1.417 |
290.2 |
|
2 b |
−15.1 |
211 |
33.1[d] |
−4.824 |
−0.954 |
293.8 |
|
5 a |
9.0 |
210 |
– |
−5.046 |
−1.384 |
291.5 |
|
5 b |
2.0 |
208 |
45.0[e] |
−4.824 |
−0.881 |
297.1 |
|
IPr |
87 |
234 |
44.5[e] |
−5.911 |
−0.480 |
270.4 |
[a] Measured for the corresponding selenium compounds. [b] Calculated at the B3LYP/def2‐TZVPP level of theory. [c] The first proton affinity (1st PA) calculated at the B3LYP/def2‐TZVPP//def2‐SVP level of theory. [d] Calculated for (2 a)CuI and (2 b)CuBr. [4c] [e] Calculated for the corresponding AuCl adducts. [19]
The Tolman electronic parameter (TEP) [20] is the most commonly used method for evaluating the electronic properties of ligands, but like other IR methods, it is not very precise and has some limitations. [21] In contrast, NMR methods usually give consistent results, which are broadly applicable.[ 3 , 4 ] The values of 31P{1H} and 77Se{1H} NMR signals of the corresponding (NHC)PPh and (NHC)Se species are equally significant but the synthesis of (NHC)Se is rather straightforward (see above).[ 3 , 4 , 22 ] The 77Se{1H} NMR data (Table 2) indicate weaker π‐accepting properties of 2 a,b, [4c] and 5 a,b compared to that of IPr. Interestingly, 5 a,b are found to be slightly more π‐acidic with respect to C5‐protonated iMICs 2 a,b. [4c] This may be attributed to the presence of an additional electron‐withdrawing aryl (Dipp) group next to the carbenic carbon atom in 5 a,b. The presence of a para‐NMe2 group in 2 b and 5 b enhances the electron density at the carbene center. Thus, 2 b/5 b are weaker π‐acceptors but stronger σ‐donors than 2 a/5 a, respectively. This trend is fully consistent with the 1 J C–Se coupling constants.[ 4a , 23 ] Smaller the value of coupling constant, larger the σ‐donor strength. Thus, a clear trend in the π‐acceptor (IPr>5 a>5 b>2 a>2 b) and σ‐donor (5 b>5 a>2 b>2 a>IPr) properties of carbenes can be concluded. The σ‐donor properties of 2 a,b, 5 a,b, and IPr can also be nicely correlated with the calculated first proton affinity (1st PA) as well as with the energy of the HOMO (Table 2). [24] The percentage buried volume (%V bur) calculated by employing Cavallo's online program SambVca[ 4f , 5a ] for 2 a,b, 5 b, and IPr is shown in Table 2. The %V bur for 5 b (45.0 %) is considerably larger than those of 2 a,b (ca. 33 %) [4c] but compares well with that of IPr (44.5 %), [19] also containing two flanking Dipp substituents.
Having analyzed the stereoelectronic properties, we decided to probe the scope of 5 a,b in palladium‐catalyzed standard C−C and C−N cross coupling reactions and compare the results with classical NHC (IPr) and iMICs (2 a,b). The corresponding PdII precatalysts were generated in situ using appropriate ligand precursors [(4 a,b)PF6, (1 a,b)Br, or (IPr)HCl], Pd(OAc)2, and a base (see the Supporting Information). For Suzuki–Miyaura C−C coupling reactions (Table S1), the yields were clearly higher with 5 a,b (Table S1, entries 4 and 5) than those with 2 a,b (Table S1, entries 2 and 3) and IPr (Table S1, entry 1). Strikingly, the amount of homocoupling byproduct was also smaller with 5 a,b (2–4 %) compared to that with IPr (6 %) and 2 a,b (8–14 %). The use of isolated compounds (IPr)(cin)PdCl and 8‐Ph (cin=cinamyl) gave comparable results (Table S2). For the C−N cross coupling of an aryl bromide and morpholine at room temperature, the use of either 5 b, 2 a, or IPr precursor gave the product in 65–99 % yield (Table S3). However, the coupling reaction of aryl chloride and morpholine with IPr (Scheme 3, entry 2) or 2 a (Scheme 3, entry 3) was rather sluggish (Scheme 3). In stark contrast, the use of 5 b gave 78–99 % conversion under similar experimental conditions (Scheme 3, entries 4 and 5), which clearly emphasizes the superior activity of 5 b over iMIC 2 a and IPr.
![Room temperature Buchwald–Hartwig C−N cross couplings.[a] GC–MS yield as an average of two independent runs.](/dataresources/secured/content-1765943094535-ac471e48-848f-40c6-b2e9-6510181cd4e1/assets/ANIE-60-2969-g005.jpg)
Room temperature Buchwald–Hartwig C−N cross couplings.[a] GC–MS yield as an average of two independent runs.
In summary, an unprecedented 1,2‐aryl migration of a series of C5‐protonated iMICs (2 a–g) under nickel catalysis to afford 1,2,4‐triayl‐1,3‐imidazoles (3 a–g) has been reported. The direct N‐methylation of 3 a–g has been shown to afford the corresponding imidazolium salts (4)X, which are precursors of the super‐iMICs (5). S‐iMIC compounds 6–8 have been isolated and characterized.
The quantification of stereoelectronic properties reveals an exceptional σ‐donor strength and steric profile (%V bur=45 %) of S‐iMICs (5), which are instrumental in steering the productivity of derived metal catalysts. As a proof of concept, this has been shown for standard C−C and C−N cross‐coupling reactions. Further studies are underway that aim to introduce S‐iMICs for more challenging chemical transformations.
Deposition Numbers 2004392, 2004393, 2004394, 2004395, 2004396, 2004397, 2004398, 2004399, 2004400, 2004401, 2004402, and 2004403 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
The authors declare no conflict of interest.
The authors gratefully acknowledge support from the Deutsche Forschungsgemeinschaft (DFG GH 129/7‐1) and thank Professor Norbert W. Mitzel for his continuous encouragement. Maurice Franke is thanked for his assistance during GC/MS analyses. The support by computing time provided by the Paderborn Center for Parallel Computing (PC2) is acknowledged. Open access funding enabled and organized by Projekt DEAL.
1
1g
1i
1i
2
2b
3
3a
3a
4
4a
4b
4c
4d
4f
5
5a
5b
6
6a
6a
6c
6d
6d
6e
6e
7
7a
7b
8
9
10
11
11a
12
12a
12b
13
13a
13b
13c
13d
14
14a
14a
14b
14b
14c
14d
15
15b
15b
15c
15c
15d
15e
15e
15g
15h
16
16a
18
19
19
20
21
22
22
23
24
24a
 Nickel‐Catalyzed Intramolecular 1,2‐Aryl Migration of Mesoionic Carbenes (iMICs)
Nickel‐Catalyzed Intramolecular 1,2‐Aryl Migration of Mesoionic Carbenes (iMICs)
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

