 Engineered SAM Synthetases for Enzymatic Generation of AdoMet Analogs with Photocaging Groups and Reversible DNA Modification in Cascade Reactions
Engineered SAM Synthetases for Enzymatic Generation of AdoMet Analogs with Photocaging Groups and Reversible DNA Modification in Cascade Reactions
Angewandte Chemie (International Ed. in English)


These authors contributed equally to this work.

- Altmetric
Methylation and demethylation of DNA, RNA and proteins has emerged as a major regulatory mechanism. Studying the function of these modifications would benefit from tools for their site‐specific inhibition and timed removal. S‐Adenosyl‐L‐methionine (AdoMet) analogs in combination with methyltransferases (MTases) have proven useful to map or block and release MTase target sites, however their enzymatic generation has been limited to aliphatic groups at the sulfur atom. We engineered a SAM synthetase from Cryptosporidium hominis (PC‐ChMAT) for efficient generation of AdoMet analogs with photocaging groups that are not accepted by any WT MAT reported to date. The crystal structure of PC‐ChMAT at 1.87 Å revealed how the photocaged AdoMet analog is accommodated and guided engineering of a thermostable MAT from Methanocaldococcus jannaschii. PC‐MATs were compatible with DNA‐ and RNA‐MTases, enabling sequence‐specific modification (“writing”) of plasmid DNA and light‐triggered removal (“erasing”).
S‐Adenosyl‐L‐methionine (AdoMet) analogs provide a way to study and control methyltransferase target sites. Their enzymatic generation has been limited to aliphatic groups at the sulfur atom. We engineered and crystallized the first SAM synthetases (PC‐MATs) able to generate AdoMet analogs with photocaging groups. In combination with DNA‐MTases, sequence‐specific modification (“writing”) of plasmid DNA and light‐triggered removal (“erasing”) is demonstrated.
Introduction
Methylation of DNA, RNA and histones is often reversible and constitutes a regulatory mechanism with direct implications in fundamental biological processes and human disease. [1] This epigenetic mark is introduced by methyltransferases (MTases), which typically use S‐adenosyl‐L‐methionine (SAM or AdoMet) as methyl donor. [2] In DNA, m5C (5‐methylcytosine) leads to inactivation of transcriptional start sites, while its oxidative removal recovers gene expression. Recently, m6A (N 6‐methyladenosine) in DNA was shown to be involved in transcriptional activation and silencing.[ 2d , 3 ] The ability to block such methyltransferase target sites and release them at a defined timepoint with an orthogonal trigger would enable the in‐depth studies required to understand their function in greater detail. [1d]
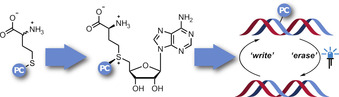
Photocaging groups are powerful tools for probing biomolecular interactions and functions. [4] They have been successfully applied for DNA, RNA, and proteins in vitro, in cells and in vivo.[ 4a , 4b ] Their removal by light reconstitutes the native biomolecule and can be controlled with excellent spatio‐temporal precision. The 2‐nitrobenzyl (ONB) group and its derivatives are widely used due to their stability and accessibility by chemical synthesis. [5] We and others recently showed that benzylic [6] and photocaged (PC) AdoMet analogs[ 5b , 7 ] are converted by MTases that are not sterically constrained and thus can be used to block the enzymes’ target sites. The action of an MTase for the installation of a PC group (“writing”) can thus be combined with light for the removal of the introduced label (“erasing”)—an important step towards investigations of epigenetic modifications with spatio‐temporal control (Scheme 1). However, this approach is limited due to the degradation of AdoMet analogs in aqueous solution, their cell impermeability and the fact that their chemical synthesis yields epimeric mixtures. [8] These issues can be circumvented by the enzymatic in situ generation of AdoMet analogs from methionine analogs using methionine adenosyltransferase (MAT) (also termed SAM‐synthetase) in vitro or in cells.[ 6a , 9 ]

Concept for enzymatic in situ generation of AdoMet analogs with photocaging groups (PC). The engineered PC‐MATs enable the reversible modification of MTase target sites, as exemplified for DNA (“writing”) starting from indicated methionine analogs (1) with photcaging (PC) groups and ATP in cascade reactions. The modifications can be removed (“erasing”) by light as an orthogonal trigger.
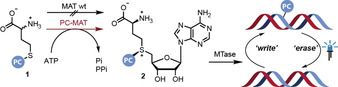
To this date, neither wildtype nor engineered MATs were reported to accept benzylic or photocaging groups,[ 6a , 10 ] although this activity would be imperative to implement this optochemical concept in a biological setting. We sought to address this shortcoming and develop MAT variants (PC‐MATs) able to efficiently convert methionine analogs bearing photocaging groups (1 a–c) to the respective AdoMet analogs (2 a–c) (Scheme 1). Such PC‐MATs should enable cascade reactions with various promiscuous MTases for the installation of photocaging groups in MTase target molecules and their light‐induced removal (Scheme 1).
Results and Discussion
For the rational re‐design of the Cryptosporidium hominis MAT (ChMAT) active site, we evaluated the crystal structure of ChMAT (PDB ID: 4ODJ). [11] The AdoMet binding pocket is formed at the interface of the two monomers (Figure 1 A), as often observed for MATs. [12] The methyl moiety of AdoMet is contained within a hydrophobic pocket, which is lined by the residues I122, C125, V126, and I330 in ChMAT (Figure 1 B). Stacking interactions between the adenine and a phenylalanine (F258) further stabilize binding of AdoMet or ATP, respectively. These features are in line with structures from other MATs that were analysed in comparison (Figure S1). Furthermore, previous characterizations of MATs revealed that a “gating loop” provides access to the active site in a dynamic manner.[ 9b , 12b , 12c , 13 ] This gating loop is formed by residues 118–130 in ChMAT (highlighted in green in Figure 1 A).
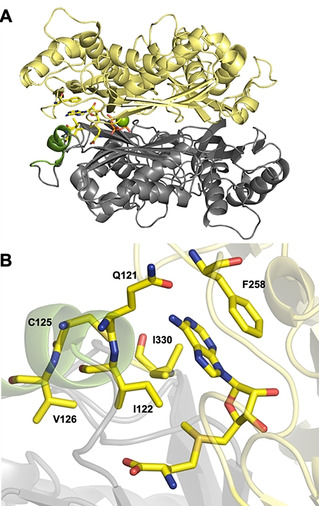
Engineering ChMAT for activity on benzyl‐ (1 d) and 2‐nitrobenzyl‐homocysteine (1 a). A) Overall structure of WT ChMAT (4ODJ). The flexible gating loop shown in green. Magnesium ions are shown as green spheres. B) Hydrophobic “pocket” surrounding the methyl moiety of AdoMet (shown as sticks). Residues I122, I330, V126 and C125 that form the pocket, as well as Q121 and F258 involved in direct interactions with the adenine moiety are also shown as sticks.
To expand the substrate scope of ChMAT towards benzylic methionine analogs, we substituted the amino acids I122, V126 and I330 (that constitute the hydrophobic binding pocket) as well as Q121 (that is part of the gating loop) by less sterically demanding residues, generating eight ChMAT variants: I122A, I122V, I122G, V126G, V126A, I330A, I330V and Q121A. These variants were tested in an enzymatic cascade reaction with Ecm1, a highly promiscuous guanine N7 methyltransferase from Encephalitozoon cuniculi that efficiently converts benzylic AdoMet analogs[ 6c , 7a , 14 ] (Figure 2).
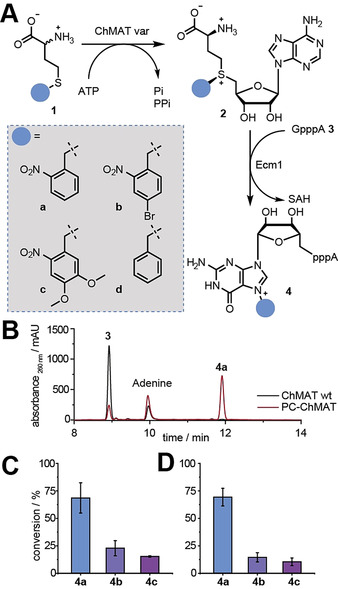
A) Enzymatic cascade reaction with ChMAT variants, Ecm1, and methionine analogs 1 a–d leading to formation of 4 a–d, respectively. B) HPLC runs for conversion of substrate 1 a in a cascade reaction with WT‐ChMAT or variant I122A/I330A (PC‐ChMAT). C,D) Conversions obtained with PC‐ChMAT (C) and triple variant ChMAT I122A/C125A/I330A (D) for indicated products. Data and error bars show average values and std. dev. of at least three independent experiments.
As expected, the WT ChMAT enzyme showed no activity on 2‐nitrobenzyl‐d,l‐homocysteine 1 a in an HPLC‐based assay (0.7±0.4 % conversion; Figure 2 B, S5). We therefore switched to benzyl‐d,l‐homocysteine 1 d as surrogate substrate, and found 5 % conversion of 1 d to the benzylated GpppA 4 d according to HPLC analysis (Table S4, Figure S4).
Testing the eight active site variants mentioned above, we found that four of them—namely I122A, I122V, I122G and V126A—displayed increased activity on 1 d compared to WT ChMAT (Table S4). Three variants (V126G, I330A, I330V) were active but did not show increased activity on 1 d. Q121A was not active on 1 d. These data suggest that positions I122 and V126 are suitable sites to increase the ChMAT activity on benzylic methionine analogs. However, combining the two beneficial mutations in double variant I122V/V126A did not further increase activity on 1 d. Thus, ChMAT I122A showed the highest activity on 1 d, yielding 25 % of 4 d. The single substitution led to a 5‐fold increase compared to the WT ChMAT. The identity of 4 d was confirmed by LC‐MS analysis (Figure S6). If the enzyme was left out, no product was formed (Figure S4).
We anticipated that the improved activity of 1 d would shift the substrate spectrum of the variants towards substituted benzylic moieties as previously observed in directed evolution. [15] Consequently, we tested ChMAT I122A (the best variant on 1 d) for activity on a methionine analog with a photocaging group at the sulphur atom. Indeed, ChMAT I122A showed activity on 1 a, yielding 5 % conversion to 4 a (Table S3). However, most of the other variants tested (I122V, I122G, the double variant I122V/V126A, Q121A nor V126A) did not result in product formation. To our delight, variant I330A yielded 23±3 % (Figure S2, Table S3) and the double variant I122A/I330A even 69±14 % of 4 a (Figure 2 B,C). Targeting an additional residue of the gating loop, did not lead to further improvements, as 69±8 % of 4 a were formed by the triple variant I122A/C125A/I330A. The formation of product 4 a was confirmed by LC‐MS (Figure S7). These three ChMAT variants were also tested on methionine analogs bearing red‐shifted ONB‐derivatives, namely 4‐bromo‐2‐nitrobenzyl‐homocysteine (1 b) and 4,5‐dimethoxy‐2‐nitrobenzyl‐homocysteine (1 c). The conversion to the respective products yielded up to 23±7 % 4 b or 15±4 % 4 c in cascade reactions according to HPLC analysis (Figure 2 C,D), suggesting that additional substituents interfere with accommodation in the active site. The respective products were confirmed by LC‐MS (Figure S8–9). The best variant I122A/I330A was termed PC‐ChMAT.
To test whether PC‐ChMAT was also compatible with other MTases, we sought to assess its utility in an enzymatic cascade reaction with a DNA MTase. We thus generated PC‐AdoMets 2 a–c in situ and coupled the reaction to a promiscuous DNA methyltransferase from Thermus aquaticus (M.TaqI), which modifies the N 6 position of adenosine, recognizing the sequence 5′‐TCGA‐3′. Subsequent irradiation by light should trigger release of the PC group, thus mimicking an epigenetic process by enzymatic writing and light‐induced erasing (Scheme 1). To this end, a short dsDNA substrate or the plasmid pBR322, respectively, was incubated with the respective methionine analogs 1 a–c, the PC‐MAT and M.TaqI, followed by enzymatic degradation to single nucleosides and LC‐MS‐based analysis. In all cases, the N 6‐modified 2′‐deoxyadenosine (6 a–c) was observed, demonstrating successful enzymatic installation of the respective PC‐group. In parallel, the identical sample was subjected to irradiation by light (365 nm for 6 a, 405 nm for 6 b,c). No modified nucleoside could be detected under these conditions, demonstrating successful and complete photodeprotection (Figures S10–12). In control experiments without enzyme, the photocaged nucleosides were not detectable either (Figures S10–12).
To understand how PC‐ChMAT (ChMAT I122A/I330A) can accommodate the ONB group of 2 a and how it demonstrates a different substrate tolerance than ChMAT I122A, we crystallized the former enzyme variant. We obtained structures of the enzyme in the apo form (6LTW, 1.65 Å) and with product 2 a bound (6LTV) at a resolution of 1.87 Å (Table S2). Compared to the WT, the overall fold of the enzyme is similar, with an r.m.s.d of 1.2 Å for apo and 0.85 Å for 2 a‐bound PC‐MAT. In apo PC‐MAT, the gating loop is disordered and we could only model the magnesium ions and two phosphates in the active site (Figure 3 A, cyan). The ligand‐bound structure was obtained by soaking of apo crystals with 2 a and PPPi (Figure 3). Here, clear electron density for both products was observed (Figure S13). The overall structure of the active site is not altered by the substitutions. Interestingly, binding of 2 a induces only partial folding of gating loop residues (118–121), which is conveyed by the hydrogen bond interactions of Q118 with the carboxyl group and a water‐mediated backbone amide interaction with the nitro group of the product (Figure S14). Because parts of the gating loop engage in crystal contacts, disorder in the gating loop might be fostered by crystal packing.
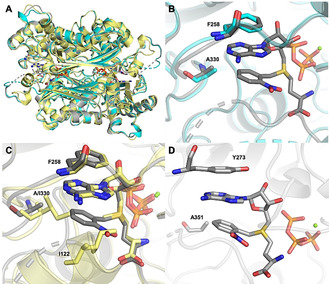
A) Superimposed structures of WT‐ChMAT (4ODJ, shown in yellow) with product bound PC‐ChMAT (6LTV, shown in grey) and apo PC‐ChMAT (6LTW, shown in cyan). B) Ligand binding by PC‐ChMAT. Residues A330, F258 (grey), PC‐AdoMet 2a (grey) and triphosphate (orange) are shown as sticks, while the Mg2+ ion is shown as sphere (green). Structure of apo PC‐ChMAT (cyan) is shown in comparison. C) Superimposed active sites of PC‐ChMAT with 2 a (grey) and WT‐ChMAT with AdoMet (yellow). Residues I122, I330, F258, ligands and triphosphate (orange) are shown as sticks, while Mg2+ ions are shown as spheres (green). PC‐AdoMet 2 a binding to WT‐ChMAT would cause steric clashes with I122 and I330. D) Modeled active sites of PC‐MjMAT (bearing equivalent mutations to PC‐ChMAT) with 2 a (grey). Residues A351, Y273, ligands and triphosphate (orange) are shown as sticks, while Mg2+ ions are shown as spheres (green).
As in WT‐ChMAT, F258 interacts with the adenine base via stacking interactions. The ONB group of PC‐AdoMet 2 a also engages in an intra‐molecular stacking interaction with the adenine ring (Figure 3 B). This conformation provides a stabilizing interaction and, at the same time, minimizes the potential for steric clashes with amino acid side chains in proximity. Indeed, the substituted amino acid A330 shows a distance of 4–4.1 Å from the 2‐nitrobenzyl ring (Figure 3 B). The superposition of the structures for the variant complexed with 2 a and the WT complexed with AdoMet shows that the substitution of residue I330 to alanine indeed decreased the steric hindrance in the active site (Figure 3 C). The nitro group of ONB would still clash with the side chain of A122 if the gating loop was closed. This suggests that its partial unfolding might support turnover of unnatural AdoMet analogs, consistent with the larger effect of the I330A substitution compared to I122A in production of 4 a (Table S3). The inverse effect was observed for production of 4 d (Table S4), which does not contain the nitro group. Here, we envision an alternative binding mode, where the gating loop is closed and substitution of I122 to alanine allows accommodation of benzyl in the active site, but I330 is required to form a tight hydrophobic pocket. These data reveal the molecular basis for the ability of PC‐ChMAT to accommodate PC‐AdoMets.
For application in different enzymatic cascades, we wanted to find out whether MATs with different temperature profiles can be engineered based on the identified substitutions. MATs from Archaea diverge considerably from bacterial/eukaryotic MATs [16] and we turned our attention to the thermostable archaeal MAT from Methanocaldococcus jannaschii. This MAT has previously been reported to be promiscuous, [17] however, no activity on methionine analogs with benzylic groups had been observed and no crystal structure of MjMAT is available. [18] Sequence alignment suggests that the substitutions L147A/I351A and L147A/V150A/I351A of MjMAT would lead to variants with similar substrate specificity as PC‐ChMAT (Figure S17). Using a homology model of MjMAT based on the crystal structure of Sulfolobus solfataricus MAT (PDB ID: 4K0B), [10b] which share a sequence identity of 51 % and are highly conserved in the active site (Figure S17), and our structure of PC‐ChMAT (PDB ID: 6LTV), we generated a model for MjMAT L147A/I351A (PC‐MjMAT) (Figure S18, 3 D). Structural analysis supports that this variant can accept 1 a–d as substrates, and we thus generated the respective MjMAT variants (Figure S19).
To assess their activity on 1 a–c, we made use of the previously described degradation of AdoMet analogs to methylthioadenosine (MTA) analogs under certain conditions, [19] including elevated temperature (Figure 4 A). As expected, the thermostable WT‐MjMAT and PC‐MjMAT converted methionine and ATP at 65 °C, leading to MTA formation (Figure S21A/B). For the methionine analogs 1 a–c, PC‐MjMAT and the triple variant were active, generating the respective MTA analogs (5 a–c), whereas WT‐MjMAT was not (Figures 4 B and S21C–J). These data show that our structural model was used successfully to design a thermostable archaeal PC‐MAT.
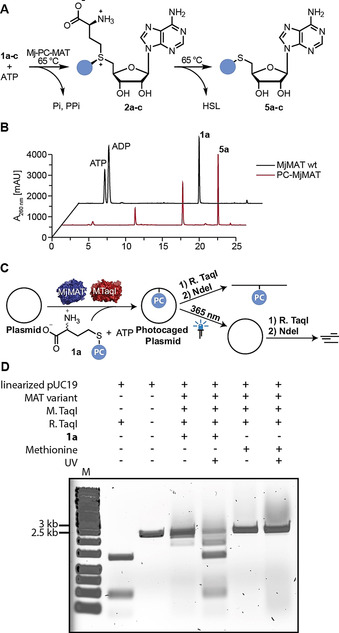
Enzymatic activity of PC‐MjMAT on 1 a and light‐reversible plasmid protection. (for 1 b,c see Figure S21). A) Assay for rapid assessment of MjMAT variants based on degradation of AdoMet at elevated temperature. B) HPLC runs for conversion of substrate 1 a with MjMAT wildtype (wt) or variant L147A/I351A (PC‐MjMAT). C) Scheme illustrating sequence‐specific modification of plasmid DNA and light‐induced removal of photocaging groups. D) Analysis of plasmid modified by PC‐MjMAT/M.TaqI cascade starting from methionine or 1 a and photo‐cleavage according to C).
We anticipated that the thermostable PC‐MAT should be ideal for cascade reactions with MTases with optimal activity at elevated temperatures, such as M.TaqI mentioned above. We used plasmid DNA containing four M.TaqI recognition sites and carried out the enzymatic cascade with PC‐MjMAT/M.TaqI starting from methionine or 1 a and ATP. If all sites are methylated or modified with the photocaging group, the plasmid is protected from restriction by the corresponding restriction enzyme R.TaqI, which recognizes the same sequence motif (Figure 4 C) and only linearization by NdeI takes place. Indeed, modification starting from methionine or 1 a led to protection of the plasmid (Figure 4 D), indicating that PC‐MjMAT efficiently generates 2 a that is readily used by M.TaqI. Irradiation of the modified plasmid did not have an effect in the case of methylation, but led to restriction by R.TaqI in the case of modification with the ONB group (Figure 4 D). Taken together these data show for the first time sequence‐specific modification of DNA (“writing”) in a MAT/MTase cascade with a photo‐caging group. Thus, sequence‐specific enzymatic writing becomes compatible with light‐triggered “erasing” starting from methionine analogs.
Conclusion
In conclusion, structure‐guided protein engineering was successful to obtain PC‐MATs, the first MAT variants reported to accept methionine analogs with photocaging groups. We showed that the PC‐methionine analogs can be efficiently utilized by different MAT enzymes, significantly expanding the substrate scope of MATs. We can now photocage plasmid DNA in a sequence‐specific manner starting from the metabolic AdoMet precursors using enzymatic MAT/MTase cascades and subsequently release it by irradiation. A detailed crystallographic analysis of the biocatalyst provides insights into the structural determinants for the expanded substrate spectrum of the variant I122A/I330A. The latter was used to model PC‐MjMAT and represents a parent for further enzyme optimization. Mutations were required to generate an enlarged hydrophobic pocket that accommodates the ONB group, which is coordinated by stacking interactions with the adenine base and hydrogen bonds of the nitro group with the gating loop backbone. With further engineering, the enzymatic cascade accepting photocaging groups bears potential for future cellular applications by blocking and releasing MTase target sites with light, which is an orthogonal trigger and provides exquisite spatio‐temporal control. This work is an important step for the implementation of in situ generated AdoMet analogs in epigenetic studies.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
A.R. thanks the ERC (772280) and the DFG (RE2796/6‐1, RE2796/7‐1 and IRTG2027) for financial support. A.O. is supported by the Graduate School of the Cells‐in‐Motion Cluster of Excellence (EXC 1003—CiM), University of Münster, Germany. D.K. thanks the DFG for funding within the Emmy‐Noether‐Program (KU2531/2). We thank Ann‐Marie Lawrence‐Dörner, Jonas Schöning, Arne Hoffmann, Stefanie Wulff and Dr. Wolfgang Dörner for excellent technical assistance. The mass spectrometry and NMR facilities of the organic chemistry department are gratefully acknowledged for analytical services. The plasmid for ChMAT herein was provided by the Seattle Structural Genomics Center for Infectious Disease (www.SSGCID.org) which is supported Federal Contract No. HHSN272201700059C from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services. The MjMAT plasmid herein was kindly provided by Prof. Jennifer Andexer (University of Freiburg). The synchrotron MX data was collected at beamline P13 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). We would like to thank Saravanan Panneerselvam for the assistance in using the beamline. Open access funding enabled and organized by Projekt DEAL.
References
1
2
2a
2a
2c
3
3a
3b
3c
4
4a
4a
4c
4d
5
5a
5b
6
6a
6a
6c
6d
7
7a
7a
7b
8
9
9a
9a
9b
10
10a
10a
10b
10c
11
12
12a
12b
12c
13
14
15
15a
15b
15b
16
18