
Oxidative addition of cyclic alkyl(amino)carbene‐coordinated phosphinidenes (MecAAC)PX to LGa affords gallium‐coordinated phosphinidenes LGa(X)‐P(MecAAC) (L=HC[C(Me)N(2,6‐i‐Pr2C6H3)]2; X=Cl 1, Br 2), which react with NaBArF 4 and LiAl(ORF)4 to [LGaP(MecAAC)][An] (An=B(C6H3(CF3)2)4 3, B(C6F5)4 4, Al(OC(CF3)3)4 5). The cations in 3–5 show substantial Ga−P double bond character and represent heteronuclear analogues of allyl cations according to quantum chemical calculations. The reaction of 4 with 4‐dimethylaminopyridine (dmap) to adduct 6 confirms the strong electrophilic nature of the gallium center, whereas 5 reacts with ethyl isocyanate with C−C bond formation to the γ‐C atom of the β‐diketiminate ligand and formation of compound 7.
Halide abstraction of L(X)GaP(MecAAC) yielded [LGaP(MecAAC][An] (An=B{C6H3(CF3)2}4; B(C6F5)4; Al{OC(CF3)3}4). The cations not only show substantial Ga−P π‐bonding contribution but can also be regarded as heteroallyl cations according to quantum chemical calculations, since the positive charge is shifted via mesomerism.
Heavier analogues of unsaturated organic molecules are intensively studied in recent years. [1] While homonuclear congeners of alkenes are well known, [2] heteronuclear group 13–15 compounds with M=E double bond (M=B−Tl; E=N−Bi), which are isovalence‐electronic to C−C bonded species, [3] are almost limited to borapnictenes with B−E (E=N, P, As) [4] and metallaimines with M−N (M=Al, Ga, In) double bonds. [5] Analogous π‐bonded heavier congeners are still rare and only one gallaphosphene, [6] three gallaarsenes [7] and five gallastibenes were structurally characterized. [8]
Heavier congeners of vinyl (C2R3 +) and allyl (C3R5 +) cations are also rare. [9] Quantum chemical calculation demonstrate that in contrast to allyl cations, which represent the prototype of π‐delocalized resonance‐stabilized carbenium cations, the conjugation in 1‐silaallyl (H2SiCHCH2 +), 2‐silaallyl (H2CSiHCH2 +), and C 2v‐symmetric trisilaallyl cations (Si3H5 +) is less effective. [10] Trisilaallyl cations and their heavier congeners therefore adopt quasi‐cyclic Cs‐symmetric structures, which provides aromatic stabilization. [11] These findings were experimentally proven with the formation of aromatic (A, B & C) [12] and homoaromatic cations (D, Scheme 1). [13] Cationic allyl‐type species (E)[ 14a , 14b ] and (F) [14c] were also reported, but the angles between the p‐orbital of the carbene ligand and the Si−Si π‐bond in E vary from 10° to 90° depending on the substituent R, whereas the p‐orbital of the carbene ligand in compound F is almost perpendicular to the Si−Si π‐bond, hence ruling out an allylic, delocalized resonance structure. Cui et al. reported on an iminoborenium cation, [15] but the p‐orbital of the carbene ligand is perpendicular to the B−N double bond. Thus, to the best of our knowledge, heteroleptic group 13/15 analogues of allyl cations are unknown, to date.

![Structurally characterized cations A–F (E: R=I, H, Me, Et); F: R=Si(i‐Pr)[CH(SiMe3)2]2); A, D: E=Si, Ge) and resonance structures of allyl and heavier homologues.](/dataresources/secured/content-1765935934664-7f82df3c-7861-4be2-8bd1-5cfcc9b5c7e0/assets/ANIE-60-1986-g004.jpg)
Structurally characterized cations A–F (E: R=I, H, Me, Et); F: R=Si(i‐Pr)[CH(SiMe3)2]2); A, D: E=Si, Ge) and resonance structures of allyl and heavier homologues.
We therefore became interested in heteroleptic allyl cation analogues. Carbene‐coordinated phosphinidenes (MecAAC)PX (X=Cl, Br) are promising precursors due to the π interaction within the P−C bond and their reactivity toward oxidative addition was demonstrated. [16] We herein report on the synthesis of complexes [LGaP(MecAAC)][An] (An=B{C6H3(CF3)2}4, B(C6F5)4, Al{OC(CF3)3}4) and their reactions with Lewis bases.
Oxidative addition reactions of LGa with (MecAAC)PX (X=Cl, Br) yielded Ga‐substituted phosphinidenes 1 and 2 (Scheme 2) in 94 % and 95 % yield, respectively. Compounds 1 and 2 are thermally stable and soluble in toluene, CH2Cl2 and THF.
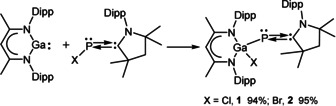
Synthesis of 1 and 2.
The 1H NMR spectra of 1 and 2 show singlets at 4.97 (1) and 5.05 ppm (2) of the γ‐CH proton as well as singlets at 1.62 ppm due to the methyl group of the β‐diketiminate backbone and three septets (1 2.67, 3.32, 3.98; 2 2.66, 3.31, 4.10 ppm) of the CHMe2 protons. The methyl groups of MecAAC exhibit single resonances at 0.85 and 1.37 ppm (1) and 0.86 and 1.45 ppm (2). While cAAC‐coordinated phosphinidenes contain the phosphinidene (major) and phosphaalkene (minor) isomers both in solution and in the solid state, 1 and 2 only form the cAAC‐coordinated phosphinidene form. The 31P NMR spectra show singlets at −21.2 (1) and −14.9 ppm (2), which are shifted to higher field compared to those of (MecAAC)PCl (161.9 ppm) and (MecAAC)PBr (146.6 ppm). [16] The 13C NMR spectra show doublets at 219.1 (1, J CP=82 Hz) and 219.4 ppm (2, J CP=84 Hz) of the carbene carbon atom, which are down‐field shifted compared to (MecAAC)PCl (210.9 ppm) and (MecAAC)PBr (210.3 ppm) but comparable with {(MecAAC)P}2SiCl2 (215.8 ppm), [17] (MecAAC)PMCl3 (M=Si, 217.4 ppm; Ge, 219.6 ppm) [18] and LSi(Cl)2P(MecAAC) (213.4 ppm, L=PhC(NtBu)2). [16]
Single crystals of 1 and 2 were obtained from toluene/n‐hexane (1) and toluene solutions (2). [19] Compounds 1 (Figure 1) and 2 (Figure S38) crystallize in the triclinic space group P .
![Molecular structure of 1 with thermal ellipsoids set at 30 % probability. All hydrogen atoms are omitted for clarity.
[19]](/dataresources/secured/content-1765935934664-7f82df3c-7861-4be2-8bd1-5cfcc9b5c7e0/assets/ANIE-60-1986-g001.jpg)
Molecular structure of 1 with thermal ellipsoids set at 30 % probability. All hydrogen atoms are omitted for clarity. [19]
The gallium atoms in 1 and 2 adopt distorted tetrahedral geometries, whereas the phosphorous atoms fall in the plane formed by C(30), C(31) and N(3). The Ga−P bond lengths (2.289 Å 1; 2.306 Å 2) are shorter compared to that of [L(Br)Ga]2PBr (2.346 Å), [20] which is attributed to the electron donating character of the carbene. The P−C bond lengths (1.736(2) Å 1; 1.7397(16) Å 2) are comparable with the major component of (MecAAC)PCl (1.75 Å), [21] as well as those reported for (CycAAC)2P2 (1.71 Å), [22] (CycAAC)PPh (1.73 Å), [23] and [PhC(NtBu)2]SiP(MecAAC) (1.73 Å), [16] respectively, but slightly elongated compared to typical P−C double bonds (1.65–1.67 Å) in non‐conjugated phosphaalkenes. [24] The Ga−P−C angles in 1 (115.1°) and 2 (115.7°) are identical. The halide substituents bound to the Ga center render 1 and 2 interesting starting reagents for subsequent reduction and halide abstraction reactions. Unfortunately, reduction reactions of 1 or 2 with KC8 and sodium naphthalene proceeded with cleavage of Ga−P bond and subsequent formation of LGa concomitant with 2,3‐diphosphabutadiene (cAAC)2P2. [22] In contrast, reactions of 1 with NaB{C6H3(CF3)2}4 and of 2 with NaB(C6F5)4 and LiAl{OC(CF3)3}4 yielded ionic compounds 3–5 (Scheme 3). Compounds 3–5 slowly decompose in CH2Cl2 solution.
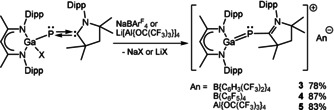
Synthesis of 3–5.
The 1H NMR spectra of compounds 3–5 show singlets at 5.85 ppm (3) and 5.86 ppm (4, 5) of the γ‐CH proton and two septets with relative intensities of 1:2 of the CHMe2 groups on both the cAAC and β‐diketiminate ligand (3 2.34, 2.77; 4 2.33, 2.77; 5 2.33, 2.77 ppm). The 13C NMR spectra show doublets at 220.1 ppm (3, 4; J CP=92 Hz) and 220.3 ppm (5; J CP=92 Hz) due to the carbene carbon atom, and the 31P NMR spectra show a peak at −56.6 ppm (3), −56.7 ppm (4) and −56.6 ppm (5), proving the neglectable influence of the different anions. Moreover, the 19F NMR spectra of 3 and 5 show singlets at −62.8 and −75.7 ppm, while that of 4 shows three resonances at −133.1, −163.8 and −167.6 ppm, corresponding to the fluorine atoms in BArF 4 and aluminate anions. In addition, the 11B NMR spectra exhibit the characteristic resonance for the boron atom in BArF 4 anion at −6.6 ppm (3) and −16.6 ppm (4).
Single crystals were grown from a toluene/fluorobenzene solution (3) and by layering n‐hexane on top of a dichloromethane solution (5). [19] Compound 3 (Figure S3) and 5 (Figure 2) crystallize in the triclinic space group P . The Ga atom adopts a distorted trigonal planar geometry (sum of bond angles 355.2°). The Ga−P bond length of 2.2393(6) Å is far shorter than the calculated single bond length of 2.35 Å [25] but longer than the calculated (2.19 Å) [26] and experimentally observed values (2.165, 2.177 Å) [6] for a Ga=P double bond. Moreover, the P−C bond length of 1.753(2) Å is slightly longer than those of 1 and 2. This reveals a resonance structure with delocalized positive charge, which is further supported by the C(30)−N(3) bond distance of 1.335(3) Å, that is typical for a double bond, as well as the significantly shorter Ga−N bond lengths of 5 compared to those of 1 and 2.
![Molecular structure of 5 with thermal ellipsoids set at 30 % probability. The highly disordered anion (see Supporting Information) and all hydrogen atoms are omitted for clarity.
[19]](/dataresources/secured/content-1765935934664-7f82df3c-7861-4be2-8bd1-5cfcc9b5c7e0/assets/ANIE-60-1986-g002.jpg)
Molecular structure of 5 with thermal ellipsoids set at 30 % probability. The highly disordered anion (see Supporting Information) and all hydrogen atoms are omitted for clarity. [19]
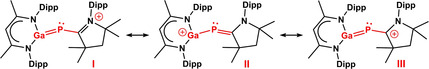
The electronic structure of the cations (3–5) was analyzed by theoretical calculations. The structures of 1 and the cationic part of 3–5 (cation G, see Scheme 4) were optimized using the approximations B3LYP [27] and B3LYP‐D3BJ [28] with the def2‐TZVP basis set. As B3LYP and B3LYP‐D3BJ rely on the same density functional, the difference between the two is a good hint to the impact of dispersion on the structure. [29]
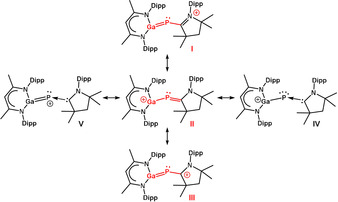
Resonance structures of the cation G. The allylic part of the mesomeric structures is indicated in red.
The structural data for 1 and cation G calculated with the B3LYP‐D3BJ method agree well with the experimental values (Table S2). To check which resonance structures (I–V in Scheme 4) best describe the cation G, NBO analyses [30] were performed and the Mayer bond orders [31] were calculated for 1 and G. If cation G can be described by the allylic mesomeric structures I–III, the transition from the neutral molecule 1 to cation G should result in a distribution of the positive charge also to C(30) and N(3) (Scheme 4) as was confirmed by the NBO charges. Following changes in the NBO charges were found: Ga(1): +0.070 e; P(1): +0.034; C(30): +0.050 e; N(3): +0.043 e. The increase of the positive charges at C(30) and N(3) is greater than that at P(1), proving that the positive charge is shifted via mesomerism. The calculated Mayer bond orders confirm the description of cation G by the mesomeric structures I–III: The bond order for the C−P bond decreases significantly going from the neutral molecule 1 (1.672) to the cation G (1.363), while the bond order for the P‐Ga bond increases (1: 1.043; G: 1.457). As the bond order for both bonds (C−P and P−Ga) amounts to 1.4 to 1.5, cation G can be considered as heteronuclear analogues of allyl cations. Calculations of a model system without sterically demanding substituents suggest that the isopropyl groups in cation G even increase conjugation in the allylic system through dispersion interaction (see Supporting Information). To investigate how sterically demanding i‐Pr groups affect the electronic structure, the model compounds S1 and SG (Figure S43) were calculated in addition to the neutral molecule 1 and cation G (data summarized in Tables S2, S3). A comparison of 1 and G versus S1 and SG calculated by means of B3LYP‐D3BJ shows, that the change in the Mayer bond orders for the transition from S1 to SG are less pronounced than the transition from 1 to cation G (Table S2), proving that the i‐Pr groups lead to an increase of the allylic resonance. The data calculated at the B3LYP level of theory show no major differences between the two systems, allowing to conclude that dispersion interaction is responsible for the increase in the resonance in cation G compared to cation SG.
To verify the electrophilic character of the Ga atom due to the strongly polarized Ga−P bond, we reacted 4 with 4‐dimethylaminopyridine (dmap). The formation of Lewis acid‐base adduct 6 proved the electrophilic nature of the gallium atom (Scheme 5).
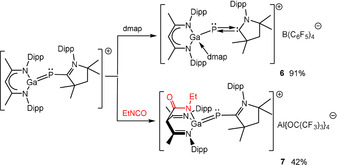
Synthesis of compounds 6 and 7 by reactions of 4 with dmap and of 5 with EtNCO (only cationic part G of 4 and 5 is shown).
Compound 6 was also synthesized by reaction of 2 with NaB(C6F5)4 in the presence of dmap. In remarkable contrast, the reaction of 5 with ethyl isocyanate EtNCO unexpectedly yielded compound 7, in which the chelating β‐diketiminate ligand is transformed into a β‐diimine ligand by electrophilic attack of the isocyanate at the γ‐carbon of the β‐diketiminate ligand backbone. Nucleophilic bond‐forming reactions involving the γ‐carbon have been previously reported for β‐diketiminate stabilized cationic transition metal [32] and Al complexes [33] as well as neutral metal complexes [34] with heteroallenes, that is, CO2, CS2, diphenylketene, iso(thio)cyanates, and other unsaturated substrates including oxygen and ethylene. [35] However, to the best of our knowledge, the formation of a tripodal N,N′,N′′ ligand as observed in compound 7 was only reported for the reaction between [LMg(n‐Bu)]2 and phenylisothiocyanate, [36] while the C−C bond forming reaction of LLi with t‐BuNCO yielded the isocyanate insertion product [{(MeCN‐2,6‐i‐Pr2C6H3)2C(CONH(t‐Bu))}Li⋅TMEDA], in which the Li cation is coordinated in an N,O‐chelating mode. [37]
The 1H NMR spectrum of 6 shows resonances of the β‐diketiminate ligand, the carbene and dmap. The singlet at 5.42 ppm (γ‐CH) is up‐field shifted compared to that of 4, and the resonance of the NMe 2 group is displayed at 3.15 ppm. The singlet at −48.7 ppm in the 31P NMR spectrum is slightly down‐field shifted compared to 4. The 1H and 13C NMR spectra of 7 prove that addition of EtNCO breaks the conjugated electronic system of the C3N2Ga ring in 5, [38] since the γ‐CH (5.36 ppm) and γ‐C (70.6 ppm) resonances are significantly up‐field shifted compared to those of 5. The 31P NMR spectrum displays a singlet at −70.7 ppm.
Single crystals of 6 and 7 were formed by layering a saturated fluorobenzene solution with n‐hexane. [19] Compounds 6 (Figure S35) and 7 (Figure 3) crystallize in the triclinic space group P .
![Molecular structure of 7 with thermal ellipsoids set at 30 % probability. The highly disordered anion (see Supporting Information) and all hydrogen atoms are omitted for clarity.
[19]](/dataresources/secured/content-1765935934664-7f82df3c-7861-4be2-8bd1-5cfcc9b5c7e0/assets/ANIE-60-1986-g003.jpg)
Molecular structure of 7 with thermal ellipsoids set at 30 % probability. The highly disordered anion (see Supporting Information) and all hydrogen atoms are omitted for clarity. [19]
The gallium atom in compound 6 adopts a distorted tetrahedral geometry with an N(4)−Ga(1)−P(1) angle of 113.91(5)°. The Ga−P bond length of 2.3202(4) Å is even longer than those of 1 and 2, which is probably attributed to the steric hindrance of dmap. Coordination of dmap weakens the π interaction of Ga−P bond, but enhances the π back donation to the carbene center. The P−C bond length of 1.7595(14) Å is comparable to those of 3–5, while the C−N bond is slightly longer as 1.35 Å. Compound 7 exhibits a bicyclo[2,2,2]octane‐like C4N3Ga core with Ga and γ‐C as joints. The Ga−P bond length of 2.2682(6) Å is slightly longer than that of 5, but shorter than the calculated single bond length of 2.35 Å, [25] while the P−C bond 1.746(2) Å falls between those observed in 5 and 1 (or 2). The Ga(1)−N(3) bond of 1.9359(17) Å is shorter than the Ga(1)−N(2) (2.0748(18) Å) and Ga(1)−N(1) (2.0609(19) Å) bonds. The C(2)−C(50) bond is longer than typical C−C single bond as 1.57 Å, and the bond distances of C(50)−N(3) (1.333(3) Å) and C(50)−O(1) (1.229(3) Å) reveal localized π electrons within NCO skeleton.
Heteronuclear congeners of allyl cations containing heavier group 13 (Ga) and group 15 (P) elements formed in halide abstraction reactions of L(X)GaP(MecAAC), which can be described as allyl cation according to quantum chemical calculations. The Ga atom in the polarized Ga=P double bond is electrophilic and reacts with strong Lewis bases, whereas nucleophilic bond‐forming reaction at the γ‐carbon atom of the ligand was observed in the reaction with ethylisocyanate.
The authors declare no conflict of interest.
Financial support from the University of Duisburg‐Essen (S.S., G.H.) is gratefully acknowledged. In memory of Professor Dietmar Seyferth (January 11, 1929–June 6, 2020), a pioneer in the field of organometallic chemistry. Open access funding enabled and organized by Projekt DEAL.
1
1a
1f
1g
1g
1j
1j
2
2a
2e
2e
3
4
4a
4a
4b
4b
4c
5
5a
5b
5b
5d
5e
5f
5f
6
6
7
7c
7c
8
8a
8b
8c
9
9a
9a
9b
10
10a
10b
10c
10d
10e
11
12
12c
12d
12f
13
13a
13b
14
14a
14b
16
16
17
18
19
20
21
22
22
23
23
24
24a
24b
24b
27
27a
27b
27c
29
30
31
32
32a
32a
32b
32c
32d
32e
32f
32g
33
33a
33b
33c
34
34b
34c
34d
34e
34f
35
35a
37
38
 Synthesis and Reactivity of Heteroleptic Ga‐P‐C Allyl Cation Analogues
Synthesis and Reactivity of Heteroleptic Ga‐P‐C Allyl Cation Analogues
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

