
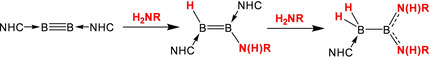
An N‐heterocyclic‐carbene‐stabilized diboryne undergoes rapid, high‐yielding and catalyst‐free hydroamination reactions with primary amines, yielding 1‐amino‐2‐hydrodiborenes, which can be considered boron analogues of enamines. The electronics of the organic substituent at nitrogen influence the structure and further reactivity of the diborene product. With electron‐rich anilines, a second hydroamination can occur at the diborene to generate 1,1‐diamino‐2,2‐dihydrodiboranes. With isopropylamine, the electronic influence of the alkyl substituent upon the diborene leads to an unprecedented boron‐mediated intramolecular N‐dearylation reaction of an N‐heterocyclic carbene unit.
Boron–nitrogen compounds have many applications in materials chemistry, hydrogen storage and medicinal chemistry. We report herein a new method for the construction of B−N bonds via the efficient and uncatalysed hydroamination of boron−boron multiple bonds with primary amines. The new diborenes generated are diboron analogues of enamines.
The metal‐catalysed hydroamination of alkenes and alkynes is a powerful and efficient method for the formation of carbon−nitrogen bonds, having grown from relative obscurity thirty years ago to become one of the most studied reactions in contemporary homogeneous catalysis. [1] In terms of thermodynamics, the addition of an N−H bond across a C=C double bond is generally thermoneutral or slightly exothermic. However, the repulsive interaction between the nitrogen lone pair and the π bond results in high kinetic barriers, and the strongly negative reaction entropy for intermolecular hydroamination reactions means that harsh thermal conditions tend to drive equilibria to the side of the starting materials. Catalysts are therefore a necessity for this chemistry, and a plethora of different systems has been developed over the past few decades. The hydroamination of alkynes [2] is also a very desirable process yielding reactive enamines and imines. [3] Despite being somewhat more thermodynamically favourable, these reactions are also plagued by high activation barriers and remain challenging.
Although clearly less well studied than their carbon isosteres, boron–nitrogen compounds are becoming established in fields such as functional polymeric materials, [4] hydrogen storage [5] and medicinal chemistry. [6] The most common routes to B−N bonds are dehydrocoupling of amine−borane adducts, salt metathesis and base‐promoted HCl elimination. Over the last few years, several protocols have been developed for the formation of B−E bonds via the addition of various polar [7] and non‐polar [8] bonds across boron−boron multiple bonds [9] in neutral, base‐stabilised diborenes and diborynes (Scheme 1). In particular, the hydrophosphination [10] of several diborenes and one diboryne to phosphinodiboranes and a phosphinodiborene, respectively, convinced us that hydroamination of such compounds was a feasible goal. To the best of our knowledge, the only reports of the non‐catalysed hydroamination of a homonuclear triple bond are reactions of primary and secondary amines with a silyl‐substituted disilyne reported by Sekiguchi and co‐workers. [11] These reactions initially produced 1‐amino‐2‐hydrodisilenes, while in one case (with pyrrolidine), a second hydroamination subsequently afforded a 1,1‐diamino‐2,2‐dihydrodisilane moiety. Herein we report the uncatalysed single and double hydroamination of a boron−boron triple bond.

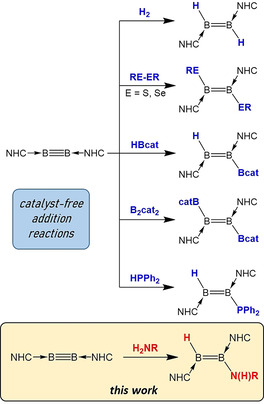
Summary of known addition reactions at boron−boron multiple bonds.
Of the neutral diborynes, LB≡BL (L=N‐heterocyclic carbene), currently known, those with saturated NHC backbones and the least steric hindrance have proved most amenable to addition reactions.[ 7c , 8b , 10 ] We therefore chose the smallest diboryne, B2SIDep 2 (1, SIDep=1,3‐bis(2,6‐diethylphenyl)imidazolin‐2‐ylidene), as a starting point for reactions with amines. Treatment with aniline in benzene solution led to a colour change from red to blue within a couple of minutes at room temperature (Scheme 2). The 11B NMR spectrum of the solution showed two new signals at 33.8 and 20.6 ppm, indicative of diborene formation. The product was isolated as a dark blue solid after workup and unambiguously identified by X‐ray diffraction as diborene 2 (Figure 1). Compound 2 has a B−B bond distance of 1.598(3) Å, which is in the expected range for diborenes, while the relatively long B−N distance of 1.520(2) Å indicates a single bond and the absence of a π interaction between these atoms. The substituents at nitrogen are arranged in a planar geometry (confirmed by DFT optimisation, see below), indicating that the N atom is sp2 hybridised with its lone pair in the remaining p orbital. This orbital is orientated perpendicular to the B=B π bond, as indicated by the B‐B‐N‐C dihedral angle of 98.2(2)°.
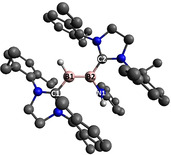
![Crystal structures of compounds 2 (above) and 7 (below) with selected atomic displacement ellipsoids at the 50 % probability level.
[18]
Hydrogen atoms other than those bound to B and N, and ethyl groups in 7, are omitted for clarity. Selected bond lengths (Å) and torsion angles (°): 2: B1‐B2 1.598(3), N1‐B2 1.520(2), B1‐C1 1.555(2), B2‐C2 1.554(2), B1‐B2‐N1‐C 98.2(2); 7: B1‐B2 1.618(5), B1‐C1 1.495(5), B2‐C2 1.593(5), B2‐N1 1.438(5), B3‐B4 1.597(5), B4‐C4 1.551(5), B4‐N2 1.516(4), B3‐C3 1.542(5) B1‐B2‐N1‐C 163.6(4), B4‐B3‐N2‐C 109.5(4).](/dataresources/secured/content-1765833687349-82ae5d2f-2cbf-47ed-b880-eafb6219bd85/assets/ANIE-60-736-g001.jpg)
Crystal structures of compounds 2 (above) and 7 (below) with selected atomic displacement ellipsoids at the 50 % probability level. [18] Hydrogen atoms other than those bound to B and N, and ethyl groups in 7, are omitted for clarity. Selected bond lengths (Å) and torsion angles (°): 2: B1‐B2 1.598(3), N1‐B2 1.520(2), B1‐C1 1.555(2), B2‐C2 1.554(2), B1‐B2‐N1‐C 98.2(2); 7: B1‐B2 1.618(5), B1‐C1 1.495(5), B2‐C2 1.593(5), B2‐N1 1.438(5), B3‐B4 1.597(5), B4‐C4 1.551(5), B4‐N2 1.516(4), B3‐C3 1.542(5) B1‐B2‐N1‐C 163.6(4), B4‐B3‐N2‐C 109.5(4).
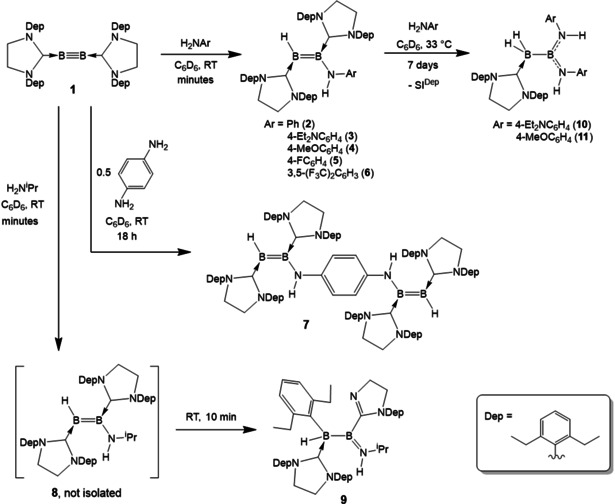
Summary of the reactivity of diboryne 1 with primary amines.
The scope of this reaction was subsequently examined by testing a series of substituted anilines, with a focus on electronic effects. The compounds 4‐methoxyaniline, 4‐diethylaminoaniline, 4‐fluoroaniline and 3,5‐bis(trifluoromethyl)aniline all reacted with 1 in a near‐identical fashion within minutes to give the corresponding 1‐amino‐2‐hydrodiborenes as blue or purple solids in high yields (84–95 %) after workup. Compounds 3 and 4, with electron‐donating nitrogen substituents, display slight low‐field shifts in the 11B NMR signal of the BN unit, while the BH boron atom is shifted to high field (e.g. for 4: δ(11B)=35.5 (BN), 18.2 (BH) ppm). For electron‐withdrawing substituents, this pattern is reversed (e.g. for 6 (3,5‐bis(trifluoromethyl)phenyl): δ(11B)=30.7 (BN), 24.8 (BH) ppm. The boron‐bound hydrogen atoms all fall within a narrow range of 2.59–2.68 ppm in the respective 1H{11B} NMR spectra, and are thus considerably more shielded than in a previously reported 1‐catecholboryl‐2‐hydrodiborene (4.27 ppm). [7c]
Comparison of the crystal structures of 2–6 [18] also revealed some interesting trends (see Supporting Information). Although marginally longer B=B bonds are observed for 3 (1.617(3) Å) and 4 (1.614(4) Å) when compared to the other derivatives (1.598(3)–1.599(2) Å), the differences fall within the experimental error. More tellingly, the B−N bonds vary in both length and the coplanarity of the substituents at B and N. In compounds 5 and 6, the N substituents are arranged such that this p orbital is practically perpendicular to the boron−boron π bond (dihedral B‐B‐N‐C for 5: −95.5(2)°; 6: −112.4(2)°). The B−N bond distances of 1.515(2) Å (5) and 1.517(2) Å (6) are at the upper end of the range for single bonds, ruling out any significant attractive π interactions. Conversely, compounds 3 and 4 have torsion angles much closer to planarity (3: −141.7(2)°; 4: −138.8(2)°), and considerably shortened B−N bonds (3: 1.465(3); 4: 1.473(3) Å).
These observations can be rationalised by comparison with well‐established enamine chemistry, where electron‐donating groups at nitrogen increase the nucleophilicity of the distal carbon atom by interaction of the nitrogen lone pair with the C=C π bond. [12] Sekiguchi and co‐workers also noted a degree of N=Si π bonding in their aminodisilene compounds, pushing electron density onto the β‐silicon atom. [11b] Increased electron density at B1 in SIDep‐supported diborenes should result in efficient B→C π bonding. Indeed, inspection of the B−C bond lengths in 2–6 supports this hypothesis. Taking compound 3 as the most prominent example, the B2−C2 distance (N‐bound boron) of 1.601(3) Å is very long and indicative of a purely dative bonding interaction. The B1−C1 bond is considerably shorter at 1.510(3) Å, and indeed shorter than all previously reported bond distances between this carbene and boron, with the exception of 1 (1.475(3) Å), and clearly reflecting significant B=C π bonding. This indicates the contribution of an N=B‐B=C butadiene‐type resonance structure (Scheme 3).
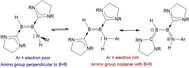
Diborene and “butadiene” structural extremes of aminodiborenes resulting from B−N rotation.
Hoping to gain further understanding of the bonding situation in 2–6, we performed DFT calculations at the B3LYP/6‐311G(d) level. As well as parent compound 2, we modelled compounds 4 and 6 as representatives of electron‐rich and electron‐poor amine substituents, respectively. Geometry optimisation reproduced the trends in bond lengths and B‐B‐N‐C torsion angles observed in the crystal structures. The frontier orbitals of 4 and 6 (Figure 2) offer an explanation for the different geometries of the molecules. The HOMOs of the two compounds largely represent the B=B π bond, as expected for diborenes, although this orbital is more localised on B1 (BH) and its associated carbene carbon atom in 4, whereas in 6 it is shared roughly symmetrically across the C‐B‐B‐C unit. The HOMO−1 varies significantly between the two compounds. In 4, the near‐coplanar orientation of the amine lone pair to the diborene unit results in a π‐bonding interaction between N and B. Conversely, in 6, the π‐symmetry lobes at nitrogen are out of phase with the B−B σ‐bonding lobes, indicating a weakly antibonding N−B interaction.
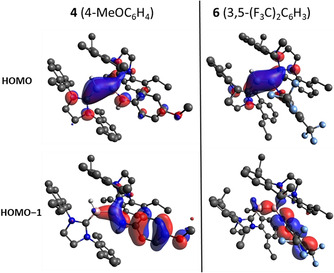
The two highest occupied molecular orbitals of aminodiborenes 4 and 6, highlighting their dependence on the aniline substituents.
The NPA natural charges of 2, 4 and 6 also depict significant variance in the electronic structures of these diborenes (Table 1). In each case, the hydrogen‐substituted boron atom (B1) bears a partial negative charge, with a positive charge of roughly equal magnitude located on amino‐substituted B2. While this leads to a relatively polar B=B bond in 2 (B1=−0.257; B2=0.238), the methoxy substituent in 4 leads to higher polarisation (B1=−0.336; B2=0.341), and the reverse is observed for 3,5‐bis(trifluoromethyl)phenyl‐substituted 6 (B1=−0.211; B2=0.202). The lower positive charge of carbene carbon atom C1 in compound 4 is also a reflection of the significant π backbonding from B1.

|
Compound (Nitrogen substituent) |
2 (Ph) |
4 (4‐MeOC6H4) |
6 (3,5‐(F3C)2C6H3) |
|---|---|---|---|
|
B1 |
−0.257 |
−0.336 |
−0.211 |
|
B2 |
0.238 |
0.341 |
0.202 |
|
C1 |
0.316 |
0.232 |
0.314 |
|
C2 |
0.294 |
0.338 |
0.285 |
|
N1 |
−0.824 |
−0.815 |
−0.796 |
Reaction of two equivalents of diboryne 1 with 1,4‐diaminobenzene gave a double hydroamination to form the first bis(diborene) compound, 7 (Scheme 2). The NMR spectra of 7 indicate a symmetrical molecule with equivalent diborene units in solution (δ(11B)=29.8, 16.1 ppm). In the solid state, however (Figure 1), the diborenes are distinct from one another, with one orientated perpendicular to the amino substituent (dihedral angle: 109.5(4)°) and the other almost coplanar (163.6(4)°). The coplanar B=B unit displays evident strong π bonding between B2 and N1 (1.438(5) Å), and B1 and C1 (1.495(5) Å), while in the other, these bonds are much longer (B3−N2=1.516(4); B3−C3=1.542(5) Å). We interpret this as evidence of electronic communication between the two diborene units. The lone pair of N2 is delocalised into the central aryl ring, which in turn acts as an electron‐rich donor towards the other diborene and causes B=N and B=C π bonding. The single set of NMR signals observed at room temperature indicates, however, that the barrier to interconversion of these states is low.
Hoping to prepare a compound with an even more pronounced butadiene‐type structure than 3 and 4, we reacted 1 with isopropylamine. Although initially the expected colour change from red to blue was observed, the solution then rapidly became yellow, with 11B NMR signals at 47.5 and −27.1 ppm. After workup and crystallisation, X‐ray diffraction revealed the compound to be diborane 9 (Figure 3), the product of a C−N activation of the target aminodiborene. The B2−N1 distance of 1.399(2) Å is that of a double bond, while the B−B distance of 1.742(2) Å is in the expected range for sp2−sp3 diboranes. [13] It appears that the further increase in electron density at B2 caused by the stronger donor can no longer be compensated for by the π acidity of the SIDep ligand, with the result that an intramolecular bond cleavage occurs, with oxidation of the boron atoms. Although several C−H activation reactions have been observed at peripheral substituents in low‐valent boron species, [14] as well as NHC ring expansion, [15] this type of N dearylation of an NHC is novel for boron compounds. A similar reaction has, however, been reported in the reduction of a boryl‐substituted N‐heterocyclic olefin, [16] and C−N activation in an NHC was recently observed in highly reducing Co(‐I) and Rh(‐I) compounds. [17] It should be noted that, whereas enamines bearing hydrogen substituents at nitrogen exist predominantly as their imine tautomers, [3b] we observed no evidence of 1,3‐hydrogen transfer from nitrogen to the distal boron atom in any of these compounds.
![Crystal structures of compounds 9 (above) and 10 (below) with selected atomic displacement ellipsoids at the 50 % probability level.
[18]
Hydrogen atoms other than those bound to B and N are omitted for clarity. Selected bond lengths (Å): 9: B1‐B2 1.742(2), B2‐N1 1.399(2), B2‐C1 1.619(2), B1‐C2 1.611(2), C1‐N2 1.291(2); 10: B1‐B2 1.726(3), B1‐C1 1.589(3), B2‐N1 1.445(2), B2‐N2 1.431(3).](/dataresources/secured/content-1765833687349-82ae5d2f-2cbf-47ed-b880-eafb6219bd85/assets/ANIE-60-736-g003.jpg)
Crystal structures of compounds 9 (above) and 10 (below) with selected atomic displacement ellipsoids at the 50 % probability level. [18] Hydrogen atoms other than those bound to B and N are omitted for clarity. Selected bond lengths (Å): 9: B1‐B2 1.742(2), B2‐N1 1.399(2), B2‐C1 1.619(2), B1‐C2 1.611(2), C1‐N2 1.291(2); 10: B1‐B2 1.726(3), B1‐C1 1.589(3), B2‐N1 1.445(2), B2‐N2 1.431(3).
Having established that diborynes can undergo uncatalysed hydroamination, we speculated that the diborenes described above may also be susceptible to further hydroamination. Indeed, gentle heating of 1 with two equivalents of the more electron‐rich anilines (p‐diethylaminoaniline, p‐anisidine) to 33 °C led, after initial rapid formation of diborenes 3 and 4, respectively, to colourless solutions after one week. In both cases, the 11B NMR signals at δ≈41 ppm and −34 ppm suggested di‐ and tricoordinate boron centres, respectively. X‐ray diffraction confirmed the formation of sp2−sp3 1,1‐diamino‐2,2‐dihydrodiboranes(5) 10 and 11 (Scheme 2, Figure 3). The compounds result from a second hydroamination across the B=B bond, providing a diaminoboryl moiety and an NHC‐stabilised dihydroboryl group. Evidently, the π basicity of the two amino groups at B2 satiates the boron atom's inherent Lewis acidity to such an extent that the NHC coordination is no longer preferred.
The yields of 10 (72 %) and 11 (80 %) are reasonably high, but the conditions required for conversion are unfortunately very specific. Attempts to shorten the reaction time by using higher temperatures resulted in significant quantities of unidentified side‐products. The less electron‐rich anilines (3,5‐bis(trifluoromethyl)aniline, 4‐fluoroaniline) do not induce a second hydroamination, while the parent aniline showed only minimal conversion after a week at 33 °C. The regioselectivity of the second hydroamination is also notable. The previously published double hydroboration [7c] of the closely related diboryne B2SIDipMes 2 (SIDipMes=1‐(2,6‐diisopropylphenyl)‐3‐mesitylimidazolin‐2‐ylidene) with catecholborane exclusively yielded the linear 2,3‐dihydrotetraborane(8). The present hydroamination reactivity can be explained by the significant partial positive charge of B2 compared to B1 (see Table 1), which is more pronounced for electron donating amines and expected to favour nucleophilic attack by the second amine molecule. This further underlines the obvious parallels to the hydroamination of disilynes by Sekiguchi and co‐workers, whose double hydroamination, which necessitated the use of a small, electron‐rich amine known to give exceptionally reactive enamines (pyrrolidine), [12b] resulted in a 2,2‐diamino‐3,3‐dihydrotetrasilane. [11b] It should also be noted that these hydroamination reactions only give isolable, identifiable products when primary amines are employed; attempts to react diboryne 1 with secondary amines such as diphenylamine and diisopropylamine provided only inseparable mixtures of compounds.
In summary, we have shown that diborynes undergo uncatalysed hydroamination reactions with primary amines to produce 1‐amino‐2‐hydrodiborenes, which can be considered boron analogues of enamines. Electron‐donating amine substituents lead to strong polarisation of the boron−boron bond, with a negative‐charge build‐up at the hydrogen‐substituted boron atom. Arylamine derivatives of these diborenes are stable and can react with a second equivalent of amine in some cases to produce 1,1‐diamino‐2,2‐dihydrodiboranes. An alkylamine derivative was found to be highly reactive and to spontaneously undergo an intramolecular N dearylation. Future work will include a full investigation of the electrochemical behavior and reactivity of the aminodiborenes, especially of compound 7, which is the first isolated example of a bis(diborene) species and thanks to its conjugated spacer should show interesting behaviour upon perturbation.
The authors declare no conflict of interest.
We gratefully acknowledge the European Research Council for funding under the European Union Horizon 2020 Research and Innovation Program (grant agreement no. 669054). We also wish to thank the Fonds der Chemischen Industrie for the award of a PhD Fellowship (to M.H.) and the Julius‐Maximilians‐Universität Würzburg for supporting this work. Open access funding enabled and organized by Projekt DEAL.
1
1a
1c
2
2a
3
3a
3b
4
4a
4a
4c
4c
4e
5
5a
5b
5d
5d
6
6a
6c
7
7a
7a
7b
7c
8
8a
8a
8b
8c
8c
8d
8d
8e
8f
8f
8g
9
9a
9a
9b
9b
10
11
11b
11c
12
12a
13
14
14a
14b
14c
14c
14e
15
15a
15a
15b
16
17
18
 Synthesis of Boron Analogues of Enamines via Hydroamination of a Boron−Boron Triple Bond
Synthesis of Boron Analogues of Enamines via Hydroamination of a Boron−Boron Triple Bond
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

