
We report the fast and selective chemical editing of ribosomally synthesized and post‐translationally modified peptides (RiPPs) by β‐borylation of dehydroalanine (Dha) residues. The thiopeptide thiostrepton was modified efficiently using CuII‐catalysis under mild conditions and 1D/2D NMR of the purified product showed site‐selective borylation of the terminal Dha residues. Using similar conditions, the thiopeptide nosiheptide, lanthipeptide nisin Z, and protein SUMO_G98Dha were also modified efficiently. Borylated thiostrepton showed an up to 84‐fold increase in water solubility, and minimum inhibitory concentration (MIC) assays showed that antimicrobial activity was maintained in thiostrepton and nosiheptide. The introduced boronic‐acid functionalities were shown to be valuable handles for chemical mutagenesis and in a reversible click reaction with triols for the pH‐controlled labeling of RiPPs.
Dehydroamino‐acid‐containing natural peptides and proteins were rapidly and selectively borylated at the β‐position using copper(II) catalysis. The introduced boronic‐acid functionalities are valuable handles for chemical mutagenesis and, in a reversible click reaction with triols, for the pH‐controlled labeling of ribosomally synthesized and post‐translationally modified peptides (RiPPs).
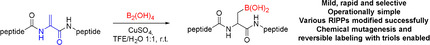
Ribosomally synthesized and post‐translationally modified peptides (RiPPs) have gained interest as alternative sources of new antibiotics due to their high activity and low development of resistance.[ 1 , 2 , 3 , 4 , 5 ] However, their clinical application is hampered by their low water solubility and poor metabolic stability.[ 6 , 7 ] Late‐stage chemical editing of these peptides is a popular approach to make semi‐synthetic analogues with improved pharmacological properties. [7] For the chemical fine‐tuning of these properties, achieving derivatization that is both chemo‐ and site‐selective is crucial. However, this is particularly challenging in the case of RiPPs due to their high structural diversity and complexity compared to generic peptides and proteins, which is a result of the post‐translational modifications occurring during their biosynthesis.
The dehydroamino acids dehydroalanine (Dha) and dehydrobutyrine (Dhb), which occur naturally in RiPPs [8] and have a distinct reactivity as carbon electrophiles, have proven to be excellent bioorthogonal handles for the selective modification of RiPPs via Michael‐type additions,[ 9 , 10 , 11 , 12 ] radical additions,[ 13 , 14 ] cross‐coupling reactions,[ 15 , 16 ] amidations, [17] cyclopropanations, [18] and cycloadditions.[ 19 , 20 ] Recently, we reported the formation of C−Si bonds in RiPPs via β‐silylation of dehydroalanines. [21] In addition, Dha residues are readily introduced chemically into proteins and have been reacted in Michael‐type additions and radical additions.[ 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 ] Methods for forming C−B bonds on Dha are currently lacking, which leaves the vast utility of such abiological functionalities unexplored in RiPPs.
Boronic acids have been applied in medicinal chemistry to act as targeting groups and to provide increased activity, stability, and water solubility,[ 31 , 32 ] which makes them interesting functional groups for the modification of RiPPs. Additionally, they are an attractive motif for side chain diversification, since they are versatile intermediates for various chemical transformations and can undergo reversible boronate ester formation with alcohols.[ 33 , 34 ] Here we report the straightforward, rapid, and selective CuII‐catalyzed β‐borylation of dehydroalanine residues in RiPPs.
The β‐borylation of α,β‐unsaturated carbonyl compounds has been reported with CuI [ 35 , 36 , 37 , 38 ] or CuII catalysis[ 39 , 40 , 41 , 42 ] and using tetrahydroxydiboron (B2(OH)4) or bis(pinacolato)diboron (B2Pin2) as the borylating agent. In contrast to the CuI‐catalyzed reactions, the CuII variant can be performed in H2O or aqueous media and open to the air, which is a promising starting point for peptide modification. To test whether Dha is a good substrate for this reaction, a protected Dha (1) was subjected to the borylation conditions reported by Santos (Scheme 1), [39] followed by oxidation to the serine derivative 2. After 24 hours, complete consumption of starting material 1 accompanied by the formation of the serine product (2) were observed in the LC‐MS of the crude product (Supporting Information, Figure S4).

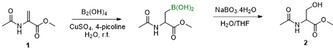
β‐borylation of Dha substrate 1. Conditions: 0.2 m 1, 1.1 equiv B2(OH)4, 1 mol % CuSO4, 5 mol % 4‐picoline, then 3 equiv NaBO3⋅4 H2O.
Encouraged by these results, we studied the β‐borylation of Dha residues in the thiopeptide thiostrepton (Figure 1 A). The conditions were slightly modified, as 2,2,2‐trifluoroethanol was used as a co‐solvent to solubilize thiostrepton. It was also found that the addition of the base 4‐picoline did not have a significant effect on the conversion, but had a negative impact on the stability of the peptide and it was therefore omitted. Full conversion of thiostrepton to one major product (3) was observed after 1 hour in the LC‐MS analysis of the crude mixture (Figure 1 B). The mass spectrum of 3 showed an m/z of 1778, which corresponds to the [M+Na]+ adduct of the doubly modified peptide, showing that thiostrepton is modified efficiently under these conditions. Singly and doubly dehydrated adducts (‐H2O) were also observed in LC‐MS and HRMS analysis of 3. Loss of water is commonly observed in the mass spectrometry analysis of borylated peptides and proteins,[ 43 , 44 , 45 , 46 ] but it is unclear whether this is the result of reversible dimerization of the boronic acids or condensation reactions with ‐OH and ‐NH2 residues in the peptide in solution, or of a dehydration reaction that occurs under the mass spectrometry conditions. However, this dehydration is fully reversible, as is evident from the follow‐up derivatization reactions of the borylated products (vide infra).
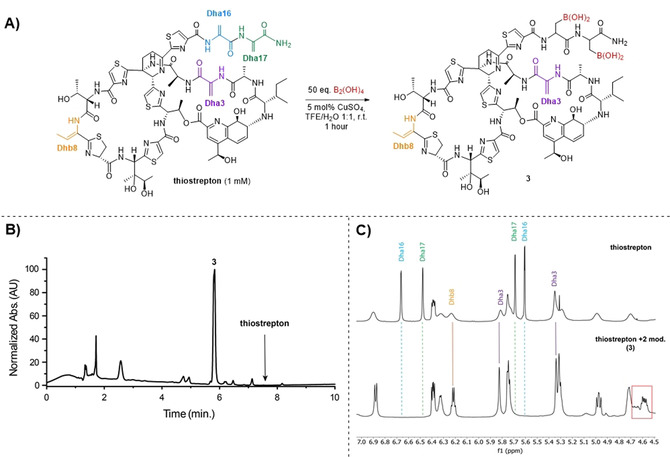
A) β‐borylation of thiostrepton (1 mm). B) LC‐MS UV chromatogram of the crude reaction mixture after 1 hour at room temperature. C) Comparison of 1H NMR spectra of thiostrepton (top) and 3 (bottom), showing the disappearance of the signals corresponding to Dha16 (blue) and Dha17 (green) and the appearance of new α‐proton signals (red box).
Product 3 was isolated as a mixture of diastereomers using preparative reversed‐phase HPLC (Supporting Information, Figure S5) and characterized by HRMS (Supporting Information, Figure S6) and 1D and 2D NMR techniques (Supporting Information, Figures S7–9).
Using 2D NMR, an authentic sample of thiostrepton was studied and the methylene signals of the different dehydrated residues were assigned (Figure 1 C, Supporting Information, Figures S7 and S8). The 1H NMR spectra of thiostrepton and 3 were then compared to show that the methylene signals of the 2 Dha residues in the tail region of thiostrepton (Dha16 and Dha17) have disappeared (Figure 1 C, Supporting Information, Figure S9). Also, 2 new signals can be observed in the α‐proton region (4.5–4.7 ppm). Together with the HRMS data, these results are consistent with a double borylation of the tail region of thiostrepton, which is known to be the most reactive part of this natural product.[ 9 , 20 ] This signifies that our method is not only rapid, but also displays a high degree of chemo‐ and site‐selectivity.
The thiopeptide nosiheptide was also subjected to the borylation conditions (Scheme 2). In this case, the addition of 4‐picoline was found to be crucial for efficient conversion to the borylated peptide. In the LC‐MS analysis (Supporting Information, Figure S10) of the crude reaction mixture 94 % conversion to the singly borylated product (4) was observed based upon integration of the different peaks in the UV chromatogram after 1 hour reaction time.
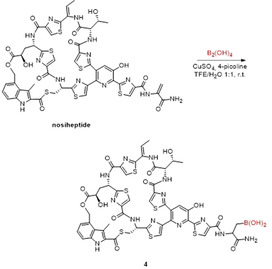
β‐borylation of nosiheptide. Conditions: nosiheptide (1 mm), 50 equiv B2(OH)4, 1.1 equiv CuSO4, 10 equiv 4‐picoline.
Next, it was investigated whether installing the boronic acid functionality on RiPPs can help to increase the water solubility; low solubility has been one of the main limitations for application of the thiopeptide class of RiPPs to which thiostrepton and nosiheptide belong. [6] 4 and nosiheptide were purified using preparative HPLC (Supporting Information, Figures S10–12) and the water solubility of thiostrepton, nosiheptide and their borylated variants in ddH2O and 50 mm TEAA (pH 7.9) was measured (Supporting Information, Figure S20). A significant (up to 84‐fold) increase in water solubility for 3 compared to unmodified thiostrepton was observed (Table 1). Due to its inherently poor water solubility, nosiheptide, as well as borylated variant 4, demonstrated a solubility that was too low to be detected (Table 1). However, the lower retention times in reversed‐phase chromatography qualitatively indicated an increased aqueous solubility also for the borylated variant of nosiheptide (4) compared to unmodified nosiheptide.

|
Antibiotic |
Solubility [μg mL−1] in ddH2O |
Solubility [μg mL−1] in 50 mm TEAA (pH 7.9) |
MIC [μg mL−1] against S. aureus |
MIC [μg mL−1] against E. faecalis |
|---|---|---|---|---|
|
Vancomycin |
– |
– |
1 |
4 |
|
Thiostrepton |
6.4±0.1 |
5.0±0.2 |
0.5 |
0.25 |
|
3 |
169.7±2.6 (27×)[a] |
422.1±8.5 (84×)[a] |
64 |
64 |
|
Nosiheptide |
N.D. |
N.D. |
0.0156 |
0.0312 |
|
4 |
N.D. |
N.D. |
0.25 |
0.5 |
[a] times more water‐soluble than thiostrepton
Furthermore, the antimicrobial activity of thiostrepton, 3, nosiheptide and 4 against two Gram‐positive strains was evaluated in a MIC assay (Supporting Information, Figure S19). The results (Table 1) show that although 3 and 4 have a decreased activity compared to the unmodified peptides, relatively high activities are preserved, especially for nosiheptide. This demonstrates that our borylation strategy can be used to improve the water solubility of RiPPs, while remaining effective against their target strains.
Having established the borylation on thiopeptides, the scope of the reaction was expanded to nisin Z, which belongs to the lanthipeptide family of RiPPs (Scheme 3 A). Its high aqueous solubility enabled performing the reaction in completely aqueous media without cosolvent. After 1 hour of reaction time at room temperature full conversion to triply borylated nisin Z was observed in LC‐MS, along with a small amount of cleavage of the two C‐terminal residues due to a known hydrolysis reaction that occurs on nisin Z in aqueous media (Supporting Information, Figure S13–15). [47] The scope was expanded further to SUMO_G98Dha, a G98Dha mutant of the 12.5 kDa protein small ubiquitin‐like modifier (Scheme 3 B). The reaction was performed in aqueous buffer and upon analysis of the reaction mixture by LC‐MS and deconvolution (Supporting Information, Figures S16 and S17) the singly borylated protein was observed. SUMO_G98Dha and the borylated product co‐eluted from the LC‐MS, indicating that the overall polarity was not affected significantly by the modification. These results show that the reaction can be applied on a wide range of challenging substrates.
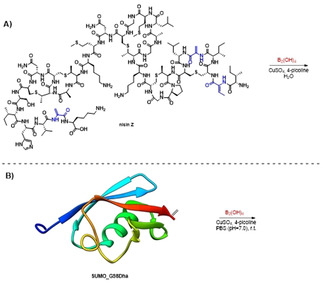
A) β‐borylation of nisin Z. Conditions: nisin Z (1 mm), 100 equiv B2(OH)4, 1.0 equiv CuSO4, 10 equiv 4‐picoline. B) β‐borylation of SUMO_G98Dha. Conditions: SUMO_G98Dha (0.1 mm), 400 equiv B2(OH)4, 10 equiv CuSO4, 40 equiv 4‐picoline.
Boronic acids are known to be useful intermediates for chemical transformations, as well as a variety of reversible reactions. Due to the post‐translational dehydration machinery, which leads to the formation of Dha and Dhb motifs, serines and threonines are highly uncommon residues in RiPPs. Since hydroboration‐oxidation is known to lead to the anti‐Markovnikov hydration of alkenes, we envisioned that chemical mutagenesis via β‐borylation of Dha residues followed by mild oxidation could be an attractive approach to re‐introduce Ser and Thr in these peptides. Purified 3 was treated with an excess amount of NaBO3 (Figure 2 A) and full conversion of the borylated residues in 3 to the Ser‐Ser motif in the tail region of thiostrepton was observed in LC‐MS (Supporting Information, Figure S21).
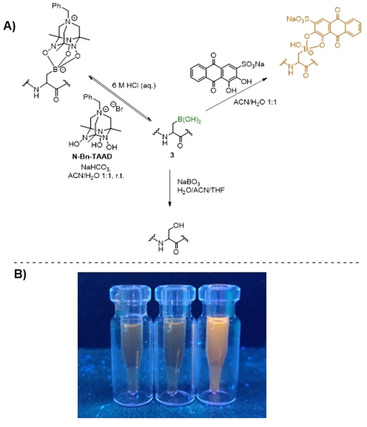
A) Oxidation of 3 with NaBO3 (conditions: 0.34 mm 3, 10 equiv NaBO3), fluorescence turn‐on labeling of 3 with Alizarin Red S (conditions: 43 μm peptide, 10 equiv Alizarin Red S), reversible formation of boronate‐triol complex (conditions: 0.58 mm 3, 25 equiv N‐Bn‐TAAD, 10 equiv NaHCO3) B) Visualization of fluorescence turn‐on of Alizarin red S. Left: DMSO control, middle: thiostrepton control, right: reaction with 3.
Installation of the boronic acid functionality onto RiPPs also allowed for the exploration of reversible boronate ester formation with alcohols. First, 3 was reacted with Alizarin Red S (Supporting Information, Figure S22), a catecholate‐based, water soluble boronic acid staining agent, which is not fluorescent on its own but becomes highly fluorescent when it forms a boronate ester (Figure 2 A). When examined under a UV light (365 nm) a clear fluorescence turn‐on effect was observed when the reaction was compared to controls containing DMSO and unmodified thiostrepton (Figure 2 B).
Boronic acids can also form boronate esters with tetraazaadamantane (TAAD) triols under basic conditions, while the resulting complexes hydrolyze readily under acidic conditions. [34] To explore this reversible labeling on RiPPs, N‐Bn‐TAAD was synthesized (Supporting Information, Figure S3) and reacted with 3 using NaHCO3 as a base (Figure 2 A). After 24 hours high conversion (>90 %) of 3 and formation of the singly‐ and doubly labeled peptide was observed in LC‐MS (Supporting Information, Figure S23). When an excess amount of aqueous HCl was added, LC‐MS showed almost full conversion back to the boronic acid within 3 hours (Supporting Information, Figure S24). After the hydrolysis the mixture was basified a second time with excess NaHCO3 and 65 % conversion to the boronate‐triol complexes was observed in LC‐MS (Supporting Information, Figure S24), proving that this labeling strategy can be applied in the reversible labeling of RiPPs under pH control. The method is also modular, since the benzyl substituent in N‐Bn‐TAAD can be switched for a variety of biologically relevant labels. The boronate‐triol coupling was also used to further confirm the presence of boronic acid in borylated SUMO_G98Dha (Supporting Information, Figures S17 and S18). Additionally, the amount of boronic acid dehydration observed in mass spectrometry analysis of the triol coupling mixtures was consistent with the extent of boronate ester formation. No dehydration was observed for products where full conversion of all boronic acids to boronate‐triol complexes was obtained, suggesting that the dehydration is reversible and showing that it does not hamper further conjugation.
Collectively, these results demonstrate that the borylation reaction presented here is a versatile method for the modification of RiPPs and proteins and it compares well with existing Dha modifications in terms of simplicity of execution, efficiency and selectivity.
In conclusion, the β‐borylation of Dha residues is a fast and selective approach for forming unnatural C−B bonds in peptides and proteins under mild conditions, allowing for the exploration of such abiological functionalities in these natural products. Installation of boronic acids on RiPPs leads to variants with improved water solubility, while antimicrobial activities are retained. Moreover, the borylated peptides have proven to be useful intermediates for further chemical transformations and reversible labeling.
The authors declare no conflict of interest.
The authors wish to thank Roos van Lier for a sample of SUMO_G98Dha and useful discussion about the manuscript and Hjalmar Permentier and Marcel de Vries for help with the HRMS analysis of borylated SUMO_G98Dha. This project was supported by the Netherlands Organisation for Scientific Research (NWO) (Vici grant 724.013.003 and ALWOP.214). G.R. acknowledges support from the Ministry of Education Culture and Science (Gravitation programme no. 024.001.035).
1
5
7
7
10
12
14
17
17
18
20
21
22
23
23
24
25
26
27
30
31
33
33
34
35
36
36
37
38
40
40
42
43
43
44
45
46
 Rapid and Selective Chemical Editing of Ribosomally Synthesized and Post‐Translationally Modified Peptides (RiPPs) via CuII‐Catalyzed β‐Borylation of Dehydroamino Acids
Rapid and Selective Chemical Editing of Ribosomally Synthesized and Post‐Translationally Modified Peptides (RiPPs) via CuII‐Catalyzed β‐Borylation of Dehydroamino Acids
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

