
The complexes [LtBuNi(OCO‐κ 2 O,C)]M3[N(SiMe3)2]2 (M=Li, Na, K), synthesized by deprotonation of a nickel formate complex [LtBuNiOOCH] with the corresponding amides M[N(SiMe3)2], feature a NiII−CO2 2− core surrounded by Lewis‐acidic cations (M+) and the influence of the latter on the behavior and reactivity was studied. The results point to a decrease of CO2 activation within the series Li, Na, and K, which is also reflected in the reactivity with Me3SiOTf leading to the liberation of CO and formation of a Ni−OSiMe3 complex. Furthermore, in case of K+, the {[K3[N(SiMe3)2]2}+ shell around the Ni−CO2 2− entity was shown to have a large impact on its stabilization and behavior. If the number of K[N(SiMe3)2] equivalents used in the reaction with [LtBuNiOOCH] is decreased from 3 to 0.5, the deprotonated part of the precursor enters a complex reaction sequence with formation of [LtBuNiI(μ‐OOCH)NiILtBu]K and [LtBuNi(C2O4)NiLtBu]. The same reaction at higher concentrations additionally led to the formation of a unique hexanuclear NiII complex containing both oxalate and mesoxalate ([O2C‐CO2‐CO2]4−) ligands.
A deceptively simple proton abstraction from nickel‐bound formate not only prepares the resulting CO2 2− unit for the release of CO in contact with electrophiles but opens up unique complex reaction schemes including reduction of nickel(II) to nickel(I) and coupling of CO2 units to give oxalate and mesoxalate. Mesoxalate formation from individual CO2 units is so far unprecedented.
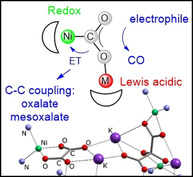
Producing chemicals and fuels from ubiquitous CO2 is a central challenge aiming at changeover towards a sustainable resource chain and much progress has been made in the last decades.[ 1 , 2 , 3 , 4 , 5 , 6 ] One general type of reactions that convert CO2 is based on an initial reductive activation step, usually occurring at a metal center, to yield in M−O(O)CIIH−, −CII(O)OH−, −CIIO2 2− or −CIIIO2 .− moieties. These species are far more reactive and can be transformed further to for example, C‐IVH4, H3C‐IIOH, H2C0O or directly to CIIO or CIII 2O4 2−. Thus, to make headway with regard to the generation of value‐added products from CO2 mechanistic insights into the transformation of reduced CO2 species are a crucial piece of information, which, however, for many systems is scarce. [7] This is, for instance, due to the fact that these intermediates are often rather elusive, so that detailed studies under controlled conditions are difficult.
Research in this direction has an impact on various different areas. As an example, in nature Ni,Fe‐carbon monoxide dehydrogenases (Ni,Fe‐CODHs) are catalyzing the reversible reduction of CO2 to CO at a redox active Ni center, which is embedded in an Fe3S4 cluster. [8] Only recently, the intermediate resulting from the initial CO2 contact was characterized crystallographically. [9] This revealed that the activation step is supported by a Lewis acidic FeII ion leading to a Ni‐C(O)O‐Fe species that features a CIIO2 2− ligand. The subsequent proceedings are still a matter of discussion. In particular it is unclear, in which step CO is liberated and what the exact role of the iron ion as a Lewis acid (LA) is. At the same time, knowledge on questions like the latter could form the basis for the rational design of artificial systems, which accomplish the LA‐assisted activation of CO2.[ 10 , 11 , 12 , 13 , 14 ]
In chemical laboratories a frequent and (especially in comparison with the CIIO2 2− dianion, sometimes referred to as carbonite) quite stable species formed in the reduction of CO2 is formate. While formate as well as its corresponding acid are important chemicals and in addition have been considered as fuels,[ 15 , 16 , 17 ] also their further conversion and thus utilization as chemical building blocks for value‐added compounds bears great potential. However, the chemistry of formate in the coordination sphere of molecular metal complexes is underdeveloped. Recently we reported on the complex [LtBuNi(OCO‐κ 2 O,C)]Li3[N(SiMe3)2]2, I, (see Scheme 1), which we synthesized from the correspondent η2‐formate precursor [LtBuNiOOCH], II, via deprotonation with Li[N(SiMe3)2]. [18] Reports on successful formate deprotonation are rather rare[ 12 , 19 , 20 , 21 ] and the synthesis of I has been the first example where it led to a mononuclear metal complex featuring a side‐on bound CO2 2− moiety; it was further shown that the same entity emerges from the activation of CO2 at LtBuNi0 units in the initial step.[ 18 , 22 , 23 ] In the present contribution we now present the results of investigations aiming at a comprehensive understanding of the properties and reactivity of the system [LtBuNi(OCO‐κ 2 O,C)]M3[N(SiMe3)2]2 with variation of the Lewis acidic metal ions M+.

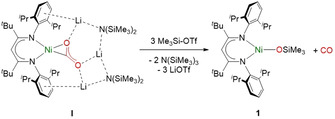
Silylation of the CO2 2− ligand in I with Me3Si‐OTf leads to the formation of complex 1 with concomitant liberation of CO.
Our studies started with gathering insights concerning the behavior of complex I in contact with electrophiles. I had proven to react with CO2 to yield [LtBuNiI−CO], III, and CO3 2−. [18] Contact with a mild acid led to [LtBuNiI−CO] (amongst others), thus resembling to some extent the conversion of CO2 2− to CO in Ni,Fe‐CODH, [8] however, the stoichiometry and redox chemistry in this transformation had remained unclear. Consequently, the first goal was to achieve a cleaner conversion of CO2 2− to CO. To accomplish this we explored, whether—instead of providing protons in the form of acids, which partially also lead to protonation of the basic [LtBu]− ligand—silylium ions can be used as proton analogues in this conversion, i.e., we studied the silylation of the CO2 2− ligand in I.
Reaction of I with three equivalents of TMS‐OTf (Scheme 1) instantly led to [LtBuNiOSiMe3] (1), which was isolated as a microcrystalline green solid in 99 % yield. A single crystal X‐ray analysis revealed, that the siloxide ligand and the [LtBu]− co‐ligand form a Y‐shaped coordination sphere for the Ni center of 1 (Figure 1).
![Molecular structure of 1.
[59]
H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.893(1), Ni‐N2 1.906(1), Ni‐O 1.772(1), O‐Si 1.607(2); N1‐Ni‐O 136.25(5), N2‐Ni‐O 127.20(5).](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g001.jpg)
Molecular structure of 1. [59] H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.893(1), Ni‐N2 1.906(1), Ni‐O 1.772(1), O‐Si 1.607(2); N1‐Ni‐O 136.25(5), N2‐Ni‐O 127.20(5).
Concomitant liberation of CO has been verified (qualitatively) via GC‐MS (21 % yield from headspace). Surprisingly, unlike in case of the reactions of [LtBuNi(OCO‐κ 2 O,C)]Li3[N(SiMe3)2]2, I, with H+ (or CO2), [18] formation of [LtBuNiI−CO], III, was not observed to occur, although TMS‐OTf likewise is an electrophilic reagent, that should react similarly. It thus may be inferred that III is formed in a secondary process, for example, through the reaction of unconsumed I with CO in the systems I/H+ or I/CO2, which is suppressed in case of TMS‐OTf, as the latter reacts with I at sufficiently high rate (Scheme 2). Indeed, we were able to show that I reacts with CO to give III (νCO=2014 cm−1, see S6). It is thus reasonable to assume that the principal reaction of I with electrophiles E+ consists of the formation of Ni‐OE units with simultaneous generation of CO (Scheme 2).
![1) Formation of CO in the reaction of [LtBuNiCO2]− with electrophiles (En+). 2) Reaction of [LtBuNiCO2]− with in situ liberated CO.](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g010.jpg)
1) Formation of CO in the reaction of [LtBuNiCO2]− with electrophiles (En+). 2) Reaction of [LtBuNiCO2]− with in situ liberated CO.
Liberation of CO from Ni centers after reduction of CO2 has been discussed quite controversially in the last years, especially with regards to the functioning of the Ni,Fe‐CODH. It is still not clear, in which step of the mechanism CO is released and what the oxidation state of the Ni center is then. Scheme 3 shows two possible mechanisms, where mechanism E1 (Scheme 3, left) involves release of CO from a NiII state with subsequent reduction of Ni before CO2 enters again and mechanism E2 (Scheme 3, right) first involves reduction to a Ni0‐CO state, followed by substitution of CO by CO2.
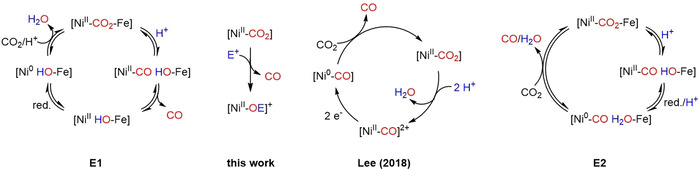
Possible pathways for CO liberation in the reaction of a Ni‐CO2 complex with electrophiles E+ (blue; for example, H+, Me3Si+).
In recent model studies with Ni‐PNP‐pincer complexes it was found that subsequent to CO2 reduction at a Ni0 center protonation of the resulting NiII−CO2 2− complexes led to NiII−CO complexes and only after consecutive reduction to Ni0 and addition of CO2, CO was released (Scheme 3, middle right circle).[ 24 , 25 ] Recent DFT studies on the enzyme also support a mechanism where CO liberation takes place only when CO2 enters the cycle (Scheme 3, E2), however, in this study Ni remained in the +1 oxidation state (not Ni0) upon reduction and CO was generated from this NiI−CO state. [26] Our findings contrast the abovementioned model studies in that CO is released directly from the NiII complex (Scheme 3, middle left) and thus supports the notion that in the Ni,Fe‐CODH reduction takes place after CO elimination or simultaneously. This matches the results of an X‐ray diffraction analysis performed for crystals of the reduced active state of CODH grown in the absence of CO, which led to a structure that was suggested as an appropriate model for this state. [27] Obviously, the ligand environment plays a crucial role in these transformations.
Bearing in mind that not many NiII‐CO complexes are known to date,[ 25 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 ] due to the limited ability of NiII to stabilize such entities via backbonding, we next tested whether a NiII‐CO complex with the LtBu− ligand system would be stable at all and reacted the cationic complex [LtBuNiII(OH2)][B(ArF)4] (2, see S3) with CO gas. A change of color from green to purple could be observed, and evaporation of all volatiles left behind a purple solid. A single‐crystal X‐ray analysis showed that indeed the H2O ligand had been replaced by CO to generate [LtBuNiII(CO)][B(ArF)4], 3, in 82 % yield (Figure 2). The structure determination further revealed that one of the aryl rings of LtBu undergoes an η2 coordination, thus completing a square planar coordination sphere of the Ni center, which also reflects in a diamagnetic 1H NMR spectrum of a solution of 3 in CD2Cl2.
![Molecular structure of 3.
[59]
H atoms and the [B(ArF)4]− counter ion are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.817(3), Ni‐N2 1.922(3), Ni‐C36 1.836(4), O‐C36 1.125(5), Ni‐C1 2.072(3), Ni‐C2 2.230(3); N1‐Ni‐C36 162.1(1), N2‐Ni‐C36 102.2(1), Ni‐C36‐O 176.6(3).](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g002.jpg)
Molecular structure of 3. [59] H atoms and the [B(ArF)4]− counter ion are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.817(3), Ni‐N2 1.922(3), Ni‐C36 1.836(4), O‐C36 1.125(5), Ni‐C1 2.072(3), Ni‐C2 2.230(3); N1‐Ni‐C36 162.1(1), N2‐Ni‐C36 102.2(1), Ni‐C36‐O 176.6(3).
Isolation of 3 shows that binding of CO to LtBuNiII is principally possible, but the initial product formed in the reaction between I and TMSOTf would be LtBuNi(CO)(OSiMe3), that is, a complex that can be derived from 3 by replacing the aryl donor in 3 by an anionic siloxide ligand with release of the counteranion. The failure to detect this product thus may suggest that binding of both CO and siloxide beside each other at the Ni center causes an unfavorable situation, leading to the formation of 1 with liberation of CO. To examine this hypothesis 3 was reacted with KOSiMe3 in CD2Cl2. The color of the solution was observed to change from purple to yellow within minutes, and the 1H NMR spectrum of the solution subsequently showed the complete absence of signals belonging to 3, while new signal sets were observed that indicated the formation of a mixture of products (see S4). One of those exhibited the characteristic signal set of 1, proving, that CO can indeed easily be replaced by ‐OSiMe3. Consistently, after removal of all volatiles a νCO stretching band ([LtBuNiI−CO]) was hardly detectable in the ATR‐IR spectrum of the product mixture, confirming that the reaction of I with TMS‐OTf leads to the generation of CO as a gas. Altogether these investigations thus show, that the contact of I with electrophiles leads to the direct formation of CO, which is of interest also considering that the CO2 2− ligand in I is derived from formate: Liberation of CO from HOOCH is known to occur in reactions with strong acids, however, there are only rather few examples, where formate coordinated to early transition metals (Ti,[ 12 , 20 ] W [21] ) releases CO upon deprotonation.
As the CO2 2− ligand in I is spanned between various metal centers (and as mentioned already can in fact also be generated by CO2 activation[ 18 , 22 , 23 ]), we were now interested in understanding the parameters that lead to activation.
Having learned that I readily generates CO in contact with electrophiles, the role of the Li3 shell surrounding the CO2 2− ligand in this conversion was explored. Initial in situ 1H NMR spectroscopic studies had indicated that the synthesis of [LtBuNi(OCO‐κ 2 O,C)]M3[N(SiMe3)2]2 is possible also with M=Na+ or K+, [18] however, no structural information had been available then. We were now able to access the Na+ derivative [LtBuNi(OCO‐κ 2 O,C)]Na3[N(SiMe3)2]2 (4) and crystallize it so that a single‐crystal X‐ray analysis could be performed, which revealed a structure that is similar to the one of I (Figure 3, left) but also shows some differences. The Ni−C bond lengths in I and 4 are almost identical (1.786(4) Å vs. 1.791(2) Å), but variations can be found for the different C−O bonds. While in 4 the bond of the C atom to the distal O atom is quite short (1.216(2) Å) and the one to the coordinated O atom is long (1.298(3) Å), for I they converge (1.234(5) Å; 1.275(5) Å). The OCO angles appear similar (127.3(2)° for 4; 128.0(4)° for I).
![Left. Molecular structure of 4⋅C7H8.
[59]
H atoms and co‐crystallized toluene are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N4 1.901(2), Ni‐N3 1.852(2), Ni‐C10 1.791(2), Ni‐O1 1.926(2), C10‐O1 1.298(3), C10‐O2 1.216(2); O1‐C10‐O2 127.3(2), Ni‐C10‐O2 157.5(2). Middle. Side view on Li3, Na3 and K3 complexes. Right. DFT‐optimized structure for the K3 adduct. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.9008, Ni‐N2 1.9711, Ni‐C1 1.8243, Ni‐O1 2.0323, C10‐O1 1.2864, C10‐O2 1.2373; O1‐C10‐O2 129.25 Ni‐C10‐O2 151.21.](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g003.jpg)
Left. Molecular structure of 4⋅C7H8. [59] H atoms and co‐crystallized toluene are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N4 1.901(2), Ni‐N3 1.852(2), Ni‐C10 1.791(2), Ni‐O1 1.926(2), C10‐O1 1.298(3), C10‐O2 1.216(2); O1‐C10‐O2 127.3(2), Ni‐C10‐O2 157.5(2). Middle. Side view on Li3, Na3 and K3 complexes. Right. DFT‐optimized structure for the K3 adduct. H atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni‐N1 1.9008, Ni‐N2 1.9711, Ni‐C1 1.8243, Ni‐O1 2.0323, C10‐O1 1.2864, C10‐O2 1.2373; O1‐C10‐O2 129.25 Ni‐C10‐O2 151.21.
It thus emerges that the nature of Lewis acidic centers around the CO2 2− unit influences its character and it was interesting to investigate how this reflects the spectroscopic properties. Hence, first of all the 13C NMR chemical shifts (δ13C) were compared for the Li3, Na3 as well as the K3 variants. A δ13C of 177.6 ppm was determined for the CO2 2− C atom in I, which shifted to 173.2 ppm upon replacement of the Li+ ions by Na+ ions, that is, in 4. A further shift to 169.5 ppm is observed for the K3‐adduct. As expected, an inverse trend was found for the νCO bands in the IR spectrum of the complexes, with frequencies of 1616 cm−1 for Li3, 1634 cm−1 for Na3 and 1637 cm−1 for the K3 adduct (1568, 1594 and 1594 cm−1 for the 13C labelled derivatives). From these data it may be inferred that the activation of CO2 is strongest in I, which is reasonable as the Li+ ions are small and hard acids interacting strongly with the O atoms, while Na+ and K+ are softer and thus pull electron density to a smaller extent. Furthermore, in 4 the CO2 appears to be somewhat more activated than in [LtBuNi(OCO‐κ 2 O,C)]K3[N(SiMe3)2]2 but the difference is not as pronounced as for the transition from I to 4.
This raised the question in how far the differences in activation translate into the reactivity of the NiCO2M3 moieties and thus also the Na+ and K+ derivatives were reacted with three equivalents of TMS‐OTf. Again, 1 was formed in both cases as verified by means of 1H NMR. However, an ATR‐IR spectrum of the solid crude product isolated after the reaction of the K+ derivative revealed that, unlike in cases of the Li+ and Na+ derivatives, additionally [LtBuNiI−CO], III, had been formed. This implicates a slower conversion in the case of K+ counterions, leaving enough time for the secondary process 2 as depicted in Scheme 2, that leads to III. While this fits to the expectations, the difference between the Na+ and K+ derivatives in the grade of CO2 activation according to IR spectroscopy is not so large, that it should lead to such a sharp contrast in reactivity. Hence, steric factors had to be considered as an origin and for this purpose the structure of [LtBuNi(OCO‐κ 2 O,C)]K3[N(SiMe3)2]2 was calculated by DFT, using the structure of 4 as a basis for an optimization after replacement of all Na+ ions by K+ ions. Comparing the resulting structure to those of I and 4 a structural transformation becomes obvious that is most pronounced in the transition from Na+ to K+ (Figure 3 middle). The trend can be well illustrated, comparing the angles between the planes spanned by the (N)2Ni(CO2) core and the plane defined by the three alkali metal cations, respectively. The small Li+ ions integrate themselves almost perfectly into the (N)2Ni(CO2) plane (3.0° deviation) and in fact even the amide N atoms almost lie in the same plane, too (N to plane distances 0.35 Å and 0.08 Å). For Na+, the twist angle between the two planes rises to 8.2°, and now the N atoms deviate from the main plane by 1.10 Å and 0.28 Å. Finally, in case of the large K+ ions the distortion becomes dramatic and the twist angle increases to 43.3°. This can clearly be seen in Figure 3, where it also becomes obvious, that in such a structure the CO2 2− unit is shielded more strongly by the SiMe3 groups of the [N(SiMe3)2]− ligands than in case of the two other derivatives, thus explaining the comparatively low reactivity.
As discussed, the contact with a Lewis acidic center can prepare a CO2 2− ligand, formed via CO2 activation at a reduced transition metal moiety, for the elimination of CO (Scheme 4). However, a M−CO2 2− moiety generated by formate deprotonation has a further option if M is redox active: an intramolecular single electron transfer leading to CO2 .−‐derived products and it seems likely that this process will also be influenced by LAs. While there is no report on a system where both pathways were accessible so far, notably, this is the case in the system described here, controlled by the incorporated Lewis acidic center, as will be outlined in the next paragraph.
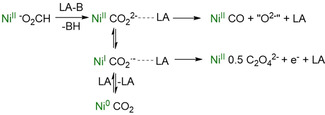
Pathways for generation and reactivity of reduced CO2 fragments with coordination of LAs.
Having shown that the nature of M+ influences the degree of activation, we hypothesized that the number of ions M+ will have an effect as well and started an investigation employing less than three equivalents of M[N(SiMe3)2]. For M=Li+ or Na+ this led to incomplete conversions to the corresponding CO2 adducts I and 4 only. While, as described above, it is possible to also generate the K3‐analogue of I and 4, namely by slow addition of [LtBuNiOOCH], II, to three equivalents of K[N(SiMe3)2], the deprotonation of II with just one equivalent of K[N(SiMe3)2] in C6D6 leads to a mixture of paramagnetic and diamagnetic products, according to a 1H NMR spectrum recorded subsequently. In order to accomplish a more uniform reaction, deprotonation was carried out then with only 0.5 equivalents of K[N(SiMe3)2] in C6D6, which proved sufficient to convert all [LtBuNiOOCH], II, employed, as evidenced by the complete disappearance of all of its signals in the 1H NMR spectrum. The latter revealed the concomitant formation of minor amounts of 1, as well as the signal sets for two paramagnetic complexes that formed as the main products. One of those was identified as the known Ni oxalate complex [LtBuNi(C2O4)NiLtBu], IV. [22] The second species shows paramagnetically broadened signals in the 1H NMR spectrum, commonly observed for NiI compounds and the presence of NiI was indeed confirmed by EPR spectroscopy, showing a rhombic signal (best simulated for a 1:1 ratio of two very similar species, Figure 4, left). Hence, under these conditions deprotonation obviously triggers an electron transfer from the CO2 2− ligand (generated by deprotonation of half of the [LtBuNiIIOOCH], II, present) to a NiII center (which presumably belongs to the second half equivalent of II, considering that all II is consumed). This would lead to a NiI species and formally a CO2 .− radical anion that likely represents the origin of the oxalate found in the second product IV. The electron transfer mentioned could either occur directly from a NiII‐CO2 2− unit, or, subsequent to an initial intramolecular electron transfer, from a NiI‐CO2 .− unit. Two equivalents of the NiII‐CO2 .− entities thus generated can then dimerize to give IV, leaving formally behind two equivalents of K[LtBuNiIOOCH], the fate of which is investigated in the following.
![X‐band EPR spectrum (26 K, 9.39 GHz) of the reaction mixture resulting from the reaction between II and 0.5 equivalents K[N(SiMe3)2] in C6D6 (black) and powder simulation for g=2.47, 2.14, 2.07 and g=2.48, 2.13, 2.08 (red). Right. EPR spectrum (26 K, 9.46 GHz) of 5 in C6D6 (black) and powder simulation for g=2.47, 2.11, 2.09 and g=2.46, 2.11, 2.06 (red).](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g004.jpg)
X‐band EPR spectrum (26 K, 9.39 GHz) of the reaction mixture resulting from the reaction between II and 0.5 equivalents K[N(SiMe3)2] in C6D6 (black) and powder simulation for g=2.47, 2.14, 2.07 and g=2.48, 2.13, 2.08 (red). Right. EPR spectrum (26 K, 9.46 GHz) of 5 in C6D6 (black) and powder simulation for g=2.47, 2.11, 2.09 and g=2.46, 2.11, 2.06 (red).
To simulate this situation, II, was reacted with KC8, which led to the formation of a vibrant red solution. Its 1H NMR spectrum exhibited a set of paramagnetically broadened signals, which indeed matched the ones observed beside the signal set of IV after the reaction with K[N(SiMe3)2]. Moreover, in the EPR spectrum a rhombic signal appeared, which was very similar to one observed before (Figure 4, right). After filtration and evaporation of the volatiles [LtBuNiI(μ‐OOCH)NiILtBu]K (5) could be isolated as a vibrant red solid in 53 % yield. X‐ray analysis of single crystals revealed a structure where two LtBuNiI moieties are bridged by a formate ligand and a K+ ion (Figure 5). Thus, the decrease of the oxidation state from +1 to +2 has led to a change of the binding mode of the formate ligand from η2 to η1 and thus to a change of the coordination sphere from square planar to trigonal planar, which is plausible. The ATR‐IR spectrum of pure 5 exhibits an asymmetric CO2 stretching vibration at 1565 cm−1 (Figure 6 c), which shifts to 1525 cm−1 when the 13C labelled formate precursor is utilized. This absorption band can also be found in the ATR‐IR spectrum of the product mixture isolated after the reaction between [LtBuNiOOCH] with 0.5 equiv. of KHMDS in C6D6 (Figure 6 a) beside the characteristic CO stretching vibration of [LtBuNi(C2O4)NiLtBu], IV (Figure 6 b). Altogether the findings lead to a stoichiometry as shown in Scheme 5 for the reaction of II with 0.5 equiv of K[N(SiMe3)2].
![Molecular structure of 5⋅2(Et2O).
[59]
H atoms and Et2O molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni1‐N1 1.861(2), Ni1‐N2 1.886(2), Ni2‐N5 1.866(2), Ni2‐N4 1.896(2), Ni1‐O2 1.894(3), Ni2‐O1 1.905(3), C1‐H1 1.03(3), K‐O1 2.672(4), K‐O2 2.681(2), C1‐O1 1.245(5), C1‐O2 1.237(6); O1‐C1‐O2 121.4(4), Ni1‐C1‐Ni2 169.6(2).](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g005.jpg)
Molecular structure of 5⋅2(Et2O). [59] H atoms and Et2O molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: Ni1‐N1 1.861(2), Ni1‐N2 1.886(2), Ni2‐N5 1.866(2), Ni2‐N4 1.896(2), Ni1‐O2 1.894(3), Ni2‐O1 1.905(3), C1‐H1 1.03(3), K‐O1 2.672(4), K‐O2 2.681(2), C1‐O1 1.245(5), C1‐O2 1.237(6); O1‐C1‐O2 121.4(4), Ni1‐C1‐Ni2 169.6(2).
![ATR‐IR spectra of the reaction mixture resulting from the reaction between II and 0.5 equiv. K[N(SiMe3)2] (a, black), pure oxalate complex IV (b, purple) and pure NiI formate complex 4 (c, red). Dotted lines indicate the correlation of the characteristic bands between a and b (1648 cm−1, dotted purple line) and between a and c (1565 cm−1, dotted red line).](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g006.jpg)
ATR‐IR spectra of the reaction mixture resulting from the reaction between II and 0.5 equiv. K[N(SiMe3)2] (a, black), pure oxalate complex IV (b, purple) and pure NiI formate complex 4 (c, red). Dotted lines indicate the correlation of the characteristic bands between a and b (1648 cm−1, dotted purple line) and between a and c (1565 cm−1, dotted red line).
![Reaction scheme for the deprotonation of LtBuNiOOCH with 0.5 equivalents of K[N(SiMe3)2].](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g013.jpg)
Reaction scheme for the deprotonation of LtBuNiOOCH with 0.5 equivalents of K[N(SiMe3)2].
Reaction of [LtBuNiI(μ‐OOCH)NiILtBu]K, 5, with one equivalent of K[N(SiMe3)2] in C6D6 does not lead to a significant decrease of the signals of 5 in the 1H NMR spectrum. Accordingly, the η2 coordination as in II appears to be a prerequisite for formate deprotonation. With this knowledge the reaction of II with more than half an equivalent of K[N(SiMe3)2] was revisited. Small amounts of K[N(SiMe3)2] were added stepwise to a solution of 13C‐labelled II and the proceedings monitored by means of 1H and 13C NMR spectroscopy (see S22). As outlined above, addition of 0.5 equiv. of K[N(SiMe3)2] led to the vanishing of all characteristic signals of II, while the signals for oxalate complex IV and NiI formate 5 reached a maximum intensity. Adding more K[N(SiMe3)2] led to the decrease of the signals of complex IV and the evolution of a new signal set that could be assigned to [LtBuNi−N(SiMe3)2] (6, see S7), until one equivalent of K[N(SiMe3)2] had been added in total. Accordingly, K[N(SiMe3)2] preferably deprotonates II, forming IV and 5, however, after completion of this conversion, K[N(SiMe3)2] displaces the oxalate ligand, with concomitant precipitation of K2C2O4. The formation of 1 as a minor side product can also be observed and may be rationalized by a minor side reaction of in situ generated 6 with traces of CO (see S8).
The formation of IV from complex II is remarkable, as the direct transformation of formate to oxalate so far has only been achieved via thermal decomposition, calcination or at elevated temperatures and high pressures from bulk CO3 2− (via formate intermediates) but is unknown in coordination chemistry.[ 40 , 41 , 42 , 43 , 44 , 45 ] There are only a few examples for oxalate formation directly from CO2[ 22 , 46 , 47 , 48 , 49 , 50 , 51 , 52 ] and one precedent case, where the reaction of formate derived CO2 2− with CO2 led to oxalate. [19]
Thus, having clarified the stoichiometry of the reaction between [LtBuNiOOCH], II, and K[N(SiMe3)2], the mechanism of oxalate formation shifted into our focus. As discussed at the end of the last but one section a conceivable intermediate is, for instance, [LtBuNiI(OCO).−]K, A, namely the product of an intramolecular electron transfer within [LtBuNiII(CO2)2−]K (see Figure 7). This does not occur within [LtBuNi(OCO‐κ 2 O,C)]K3[N(SiMe3)2]2 as there two further K+ ions pull electron density. As many attempts to trap an intermediate failed, we pursued information through computational studies and modelling of the binding situation in this species. As the starting structure for a geometrical optimization of intermediate A we used the structural data of [LtBuNiINCO]K (8, see S9), which is isoelectronic with a NiI(CO2) complex, and exchanged the cyanate ligand by CO2 .−. The resulting structure (Figure 7, left) is stable with a NiI ion binding a bent CO2 .− ligand (135.8°; 134° for CO2 .−[53]), supported by a K+ ion, which interacts with the C atom and the Ni bound O atom of the CO2 .− ligand. A very similar coordination mode with an exposed C atom has been proposed as the initial intermediate for CO2 reduction to oxalate catalyzed by Cu complexes. [47] In the system discussed here, intermediate A would transfer an electron (and K+) to unconsumed II, forming half an equivalent of IV.
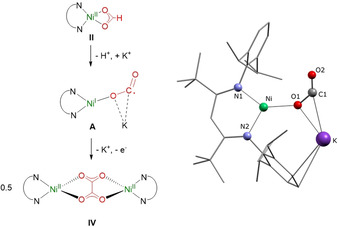
Left. Possible intermediate A leading to oxalate formation. Right. DFT‐optimized structure for A. H atoms are omitted for clarity.
In order to quantify the formation of the oxalate complex, crystallization attempts from benzene and hexane were conducted. In case of benzene (ca. 0.03 M) this gave solely purple crystals of the oxalate complex [LtBuNi(C2O4)NiLtBu] (IV, 12 % absolute yield, 48 % relative yield). However, when working at high concentrations (ca. 0.15 M), from hexane solutions repeatedly and reproducibly also small orange crystals grew, that is, a further product was generated at high concentrations. Extraction with Et2O and subsequent crystallization led to the enrichment of the orange crystals (see S10) and single‐crystal X‐ray crystallographic analysis revealed the structure of the unique multinuclear complex [(LtBuNi)6(C2O4)2(C3O6)2K6] (9, Figure 8). The quality of the crystals did not allow an anisotropic refinement of all atoms during the solution of the structure, and hence the latter does not permit a discussion of metric parameters, but the molecular structure determined and the constitution are without doubt. 9, which was formed in up to 9 % yield based on formate (6 % based on LtBuNi), contains, as IV, oxalate ligands, but also ligands with the configuration C3O6. Accordingly, three formate‐derived CO2 units have been coupled, which to our knowledge has never been reported so far. However, while the formation of oxalate (C2O4 2−) from two CO2 .− species (vide supra) is straight forward, a threefold coupling would give the unknown radical C3O6 .3−. Therefore, in a more realistic scenario two CO2 .− equivalents and one CO2 2− entity are combined yielding in the fourfold deprotonated form of dihydroxymalonic acid (C3O6 4−), which indeed is known as the hydrate of mesoxalic acid and represents a stable compound; for its formation routes via reaction of oxalate with CO2 2− or of a C2O4 .3− intermediate with CO2 .− are conceivable. 9 then contains two quadruply charged mesoxalate and two doubly negatively charged oxalate anions as well as six [LtBu]− ligands. This leads to 18 negative charges which are compensated by six K+ ions and six NiII ions, partly with tetrahedral and partly with square planar coordination spheres, which is reasonable (note that the oxalate complex IV has tetrahedrally coordinated Ni centers, while II features a square planar coordination).
![Left: Molecular structure of 9⋅2 Et2O.
[59]
Atoms that could not be refined as ellipsoids are depicted as balls. Middle. Formate and CO2 and C−C coupling products. Right: Ball and Stick structure of the core of 9⋅2 Et2O. H atoms, Et2O molecules and [LtBu]− C atoms are omitted for clarity.](/dataresources/secured/content-1765942900421-56794aad-1500-4abc-aa76-ff5c5ef84c27/assets/ANIE-60-2312-g008.jpg)
Left: Molecular structure of 9⋅2 Et2O. [59] Atoms that could not be refined as ellipsoids are depicted as balls. Middle. Formate and CO2 and C−C coupling products. Right: Ball and Stick structure of the core of 9⋅2 Et2O. H atoms, Et2O molecules and [LtBu]− C atoms are omitted for clarity.
Separation of 9 from residual IV proved possible by extraction with MeCN that dissolves 9 to give a clear orange solution, from which an orange solid was isolated. However, storage of such solutions for crystallization leads to the precipitation of a white solid, indicating a certain instability of IV in MeCN. Thus, all analytical methods had to be applied within a short period of time after contact with MeCN.
The ATR‐IR spectrum of 9 shows bands at 1666, 1617, 1588 and 1572 cm−1 in the νCO region, originating from the different carbonyl groups in the oxalate and mesoxalate ligands (1648 cm−1 for IV).
The 1H NMR spectrum displays a combination of diamagnetic and paramagnetically shifted signal sets, as one would expect due to the different coordination environments of the six Ni centers (four square planar; two tetrahedral) in the crystal structure. While a detailed assignment of the signal sets is not possible, the ratio of the integrals from paramagnetic to diamagnetic signals should be around 1:2 or less (depending on how perfect the square planar coordination modes are maintained in solution), matching the 1:1.6 found for solutions of IV in [D3]acetonitrile.
In the 13C NMR spectrum of MeCN‐d3 and C6D6 solutions of 9 neither characteristic signals for oxalate nor for mesoxalate could be detected (the commercially available dihydroxymalonic acid disodium salt resonates at 171 and 91 ppm in D2O), not even after 13C‐labelling. This is not surprising as also the corresponding 13C NMR signal of the oxalate ligand in IV eluded detection due to its connection with the paramagnetic NiII centers. However, trying to study the in situ formation of 9 through deprotonation of 13C II in hexane‐d14 or C6D6 in an NMR tube sealed with a J. Young valve, after 4 days a characteristic set of signals at 177.7 ppm (dd, 1JCC=57.8 Hz) and 103.9 ppm (t, 1JCC=57.8 Hz) could be detected (see S10; compare: 1JCC of 1,2–13C oxalic acid diethyl ester is 58.5 Hz). This consolidates the formation of mesoxalate, and its detection by 13C NMR spectroscopy in this experiment likely became possible, as 9 is not stable in solution over four days, so that the ligands get detached from the paramagnetic Ni centers.
Mesoxalate is found rather rarely in coordination chemistry. There are reports of Pd and Cu complexes with mesoxalate ligands, generated through decomposition (after heating/time) of D‐erythrulose (C4H8O4; Pd) [54] or D‐glucuronate (C5H8O6; Cu). [55] Although it does not concern mesoxalate directly, a publication by Meyer and co‐workers is noteworthy in this context, too, as it describes the reaction of CO2 with a coordinated diketone‐derived enolate (an oxalate equivalent) and based on this finding they envisioned that the coupling of three CO2 building blocks should be possible. [56] The formation of 9, which involves the reduced CO2 species CO2 2− is to our knowledge the first experimental example of a direct threefold coupling of CO2 entities, illustrating the feasibility of such transformations. This may even be relevant to the prebiotic synthesis of organic compounds,[ 57 , 58 ] and motivates further attempts to synthesize multi‐carbon compounds directly from formate or CO2.
In summary, we have shown that in complexes of the type [LtBuNi(OCO‐κ 2 O,C)]M3[N(SiMe3)2]2 (M=Li, Na, K), formed via deprotonation of the formate complex with corresponding metal amides, the CO2 2− ligand is prepared for facile CO elimination, which is triggered by a contact with electrophiles and influenced by the nature of the Lewis acidic alkali metal cations. Investigating the formate deprotonation with 0.5 equivalents of K[N(SiMe3)2] we could demonstrate for the first time the oxidative coupling of formate on defined Ni sites. This yields not only in 0.25 equivalents of oxalate, but also in 0.5 reducing equivalents, leading to the formation of a dinuclear NiI formate complex, which explains why substoichiometric amounts of K[N(SiMe3)2] proved sufficient to consume all formate precursor. A simple deprotonation reaction thus induces Ni reduction and the formation of a C−C coupling product.
Altogether, formate deprotonation can give rise to reduced CO2 species, just as the direct reduction of CO2 does, and can thus lead to the same products (CO, oxalate). However, the simplicity of a deprotonation reaction with solid precursors (instead of using gaseous CO2) and the easy variation of counter ions makes this route very convenient for systematic analysis of reduced CO2 species. Additionally, we were able to show that the same deprotonation reaction conducted at higher concentrations can even lead to mesoxalate, formed in an unprecedented, direct threefold coupling of reduced CO2 units.
Hence, in summary, the combination of a Ni formate with an amide base that deprotonates and at the same time introduces Lewis acidic centers generates a versatile system that bears a lot of potential, aiming at the further utilization of formate or CO2 to generate more complex organic molecules. Both can be employed to generate a CO2 2− species at a metal center and the interaction with a LA can decide upon the proceedings. If they are strong the entity is stabilized but activated for CO elimination in contact with an electrophile, if it is weak, the positioning of the electrons can be shifted towards the metal (this of course depends on the properties of the metal), which opens up CO2 .− chemistry. Here, coupling reactions have been studied but a trapping by other substrates is conceivable. Hence, the development of CO2 reduction catalysts (or such that utilize formate) in this direction requires a fine balancing and (for a given central metal) testing of a variety of LAs.
The authors declare no conflict of interest.
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008/1–390540038. Open access funding enabled and organized by Projekt DEAL.
1
2
3
5
7
9
9
12
13
14
15
16
18
18
19
19
20
21
22
29
31
32
33
34
35
36
37
37
38
39
40
41
42
43
45
47
49
50
51
52
52
53
56
56
57
58
59
 Selective Transformation of Nickel‐Bound Formate to CO or C−C Coupling Products Triggered by Deprotonation and Steered by Alkali‐Metal Ions
Selective Transformation of Nickel‐Bound Formate to CO or C−C Coupling Products Triggered by Deprotonation and Steered by Alkali‐Metal Ions
 Facebook
Facebook
 Twitter
Twitter
 Linkedin
Linkedin
 Whatsapp
Whatsapp

