 Design, Identification, and Evolution of a Surface Ruthenium(II/III) Single Site for CO Activation
Design, Identification, and Evolution of a Surface Ruthenium(II/III) Single Site for CO Activation
Angewandte Chemie (International Ed. in English)



- Altmetric
RuII compounds are widely used in catalysis, photocatalysis, and medical applications. They are usually obtained in a reductive environment as molecular O2 can oxidize RuII to RuIII and RuIV. Here we report the design, identification and evolution of an air‐stable surface [bipy‐RuII(CO)2Cl2] site that is covalently mounted onto a polyphenylene framework. Such a RuII site was obtained by reduction of [bipy‐RuIIICl4]− with simultaneous ligand exchange from Cl− to CO. This structural evolution was witnessed by a combination of in situ X‐ray and infrared spectroscopy studies. The [bipy‐RuII(CO)2Cl2] site enables oxidation of CO with a turnover frequency of 0.73×10−2 s−1 at 462 K, while the RuIII site is completely inert. This work contributes to the study of structure–activity relationship by demonstrating a practical control over both geometric and electronic structures of single‐site catalysts at molecular level.
An air‐stable surface [bipy‐RuII(CO)2Cl2] single site is designed towards CO oxidation, while all other RuIII single sites are not active. The methodology is further extended to nine transition‐metal single‐site systems, enabling the use of surface coordination chemistry in heterogeneous catalysis.
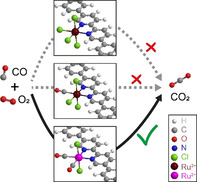
Introduction
RuII complexes were first discovered in the 19th century. [1] Their chemistry is diverse and has sparked wide interest in homogeneous catalysis, [2] photocatalysis, [3] and biochemical and biomedical applications. [4] Most RuII compounds function as reducing agents and only a few RuII complexes with strong‐field π‐acceptor ligands are air‐stable.[ 1 , 5 ] The standard reduction potentials E ⊖ of RuIII/RuII in [Ru(H2O)6]3+/ [Ru(H2O)6]2+, [Ru(NH3)6]3+/ [Ru(NH3)6]2+, [Ru(en)6]3+/ [Ru(en)6]2+, [Ru(CN)6]3−/ [Ru(CN)6]4− and [Ru(bipy)6]3+/ [Ru(bipy)6]2+ pairs are 0.23 V, 0.10 V, 0.21 V, 0.86 V and 1.24 V, respectively. [6] Nonetheless, the RuII system can be oxidized by molecular O2 at elevated temperatures, limiting its study and application in heterogeneous oxidation chemistry.
Inspired by the relatively high reduction potential of [Ru(bipy)6]3+/[Ru(bipy)6]2+ pair, we covalently bind bidentate bipyridine (bipy) ligand into the polyphenylene (PPhen) framework via a C−C coupling reaction, converting the bipy ligands from solute molecules into surface binding sites. This enables the design of surface single‐site and single‐atom catalysts, which mimic molecular catalysts and have unique coordination and electron configuration compared with particles and clusters. [7] These reported single sites, usually supported by metal oxide or carbon, have shown unique activity or selectivity in the water‐gas‐shift reaction, [8] hydrochlorination, [9] methane activation, [10] CH3OH steam reforming, [11] and hydrogenation. [12] In this work, we successfully designed and prepared the surface [bipy‐RuII(CO)2Cl2] single site via simultaneous RuIII to RuII transition and Cl−/CO ligand exchange. Such solid/gas interface coordination chemistry, including both geometric and electronic structures, is monitored by a combination of in situ X‐ray absorption fine structure (XAFS), diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), ex situ far‐FTIR, X‐ray photoelectron spectroscopy (XPS), and density functional theory (DFT) calculations. The [bipy‐RuII(CO)2Cl2] site is stable in air for at least 400 days. It enables CO oxidation with a turnover frequency (TOF) of 0.73×10−2 s−1 at 462 K, while the RuIII site is completely inert. Such distinct catalytic performance could result from the geometric configuration of the [bipy‐RuII(CO)2Cl2] site and its optimized energy level that boost CO conversion.
Building a surface single site on PPhen with bipy ligands is proven to be a general and versatile method. Eleven types of uniform and well‐defined surface single sites are obtained, including [bipy‐FeCl4]−, [bipy‐CoCl2], [bipy‐Ni(H2O)4]2+, [bipy‐CuCl2], [bipy‐Cu(H2O)4]2+, [bipy‐ZnCl2], [bipy‐RuCl4]−, [bipy‐RuBr4]−, [bipy‐RhCl4]−, [bipy‐PdCl2], and [bipy‐PtCl4]. This variety of selection including both metal cations and ligands suggests great potential in catalysis and mechanistic understandings.
Results and Discussion
Design and determination of the [bipy‐RuIIICl4]− single site.
Our strategy utilizes Suzuki cross‐coupling with 5,5′‐dibromo‐2,2′‐bipyridine, 1,2,4,5‐tetrabromobenzene, and benzene‐1,4‐diboronic acid as the building blocks (Figure 1 a). The latter two comprise the backbone of the PPhen framework [13] while 5,5′‐dibromo‐2,2′‐bipyridine provides bipy ligand functionality. We choose PPhen as the backbone for its pure aromatic structure, microporosity and high thermal stability under air (up to 400 °C), as reported previously. [13] Bipy is a typical chelating ligand that can stabilize metal cations. [14] It has been functionalized within the UiO‐67 [15] and polystyrene system recently. [16] The ratio between 5,5′‐dibromo‐2,2′‐bipyridine and 1,2,4,5‐tetrabromobenzene determines the amount of bipy ligands in relation to the PPhen‐bipy polymer, thereby the loading of single metal sites. The ratio is set to 1:3 for that raising the content of dibromo molecule will increase the linear content of the polymer, decrease the overall surface area and consequently reduce the accessibility to the bipy sites. [17] The PPhen‐bipy is Pd free, as shown in the energy dispersive spectroscopy (EDS; Supporting Information, Figure S1).

![Identification of [bipy‐RuIIICl4]− single site. a) The general approach in creating the single‐site coordination environment. b) HAADF‐STEM images of PPhen‐[bipy‐RuIIICl4]H. c) Ru K‐edge XANES spectra of PPhen‐[bipy‐RuIIICl4]H, Ru foil, RuCl3, and RuO2. d) Experimental and fitted EXAFS data of PPhen‐[bipy‐RuIIICl4]H. The inlet is the DFT simulation of the [bipy‐RuIIICl4]− structure.](/dataresources/secured/content-1765837351133-55816efd-9798-4b19-954a-9e67aead3496/assets/ANIE-60-1212-g001.jpg)
Identification of [bipy‐RuIIICl4]− single site. a) The general approach in creating the single‐site coordination environment. b) HAADF‐STEM images of PPhen‐[bipy‐RuIIICl4]H. c) Ru K‐edge XANES spectra of PPhen‐[bipy‐RuIIICl4]H, Ru foil, RuCl3, and RuO2. d) Experimental and fitted EXAFS data of PPhen‐[bipy‐RuIIICl4]H. The inlet is the DFT simulation of the [bipy‐RuIIICl4]− structure.
The PPhen‐bipy reacts with RuCl3 to form a [bipy‐RuCl4]− single site in the presence of HCl, in which the bipy ligand anchors the Ru3+ while Cl− anions are balancing the +3 charge. High density and good dispersion of single sites are achieved, with more than 200 Ru ions identified over 40 nm2 of high angle annular dark field‐scanning transmission electron microscopy (HAADF‐STEM) image (Figure 1 b). The loading of Ru and Cl is confirmed in the EDS and X‐ray photoelectron spectroscopy (Supporting Information, Figures S2, S3). The absorption edge position of Ru K‐edge X‐ray absorption near‐edge structure (XANES) is almost identical to RuCl3 rather than Ru0 foil or RuO2, suggesting that the oxidation state is RuIII (Figure 1 c). The extended X‐ray absorption fine structure (EXAFS) further confirms the single‐site nature, showing only Ru−N and Ru−Cl scattering with no Ru−Ru scattering (Figure 1 d). The coordination numbers and bond lengths of Ru−N and Ru−Cl scattering are 2.0 at 2.05±0.02 Å and 4.0±0.4 at 2.35±0.01 Å, respectively (Supporting Information, Table S1). The calculated bipy‐RuIIICl4 − structure using DFT (Figure 1 d inlet; Supporting Information, Figure S4a) shows very similar values of Ru−N at 2.096 Å and Ru−Cl at 2.400 Å (Supporting Information, Table S2). The bond lengths in both EXAFS and DFT calculations match the reported value of molecular K[bipy‐RuIIICl4] complex (2.36 Å for Ru−Cl and 2.02 Å for Ru−N; Supporting Information, Table S3 entry 18). [18] The PPhen‐[bipy‐RuIIICl4]− is stable in air up to 350 °C (Supporting Information, Figure S5), suggesting its capability for reactions at elevated temperature even in oxidative environments. The 5.8 wt % Ru loading is calculated from the difference in weight residue between the PPhen‐[bipy‐RuIIICl4]− and PPhen‐bipy. The value is smaller than the circa 8.6 wt % Ru loading calculated from the ratio of the bipy ligand in the PPhen‐bipy (see calculation in Supporting Information). The possible reasons are the loss of bipy during PPhen‐bipy synthesis and the formation of inaccessible bipy site. The BET equivalent surface area is 465 m2 g−1, providing the porous framework for heterogeneous catalysis (Supporting Information, Figure S6 and Table S4). Obtaining such well‐defined [bipy‐RuIIICl4]− site is crucial for determining the solid/gas interface coordination chemistry, which provides the possibility to trace further structure evolution.
Structural Evolution from [bipy‐RuIIICl4]− to mer(Cl)‐[bipy‐RuIII(CO)Cl3]
H2 reduces [bipy‐RuIIICl4]− into Ru nanoparticles (Supporting Information, Figure S7), owing to the fact that H2 takes away Cl‐ and no other ligand is available at the surface to maintain the hexacoordinated octahedral structure. Consequently, CO, as a reductive gas phase ligand, is selected for this ligand exchange reaction at the solid/gas interface. O2 is co‐fed, thereby any detection of the RuII will guarantee its stability under molecular O2. At room temperature, the initial [bipy‐RuIIICl4]− remains unchanged (Supporting Information, Figure S8), showing no ligand exchange with CO. The release of HCl is observed starting from 433 K (Figure 2 a). A surface adsorbed C≡O vibration is found in the DRIFTS spectra at 2057 cm−1 (Figure 2 b). This vibration absorption corresponds well to the FTIR spectrum of molecular mer(Cl)‐[Ru(bipy)(CO)Cl3] (Supporting Information, Table S5 complex D). [19] The fitted EXAFS spectrum shows the decrease of Ru−Cl coordination number from 4.0±0.4 to 3.2±0.2 while maintaining the bond distance at 2.35±0.01 Å (Figure 2 c; Supporting Information, Figures S9b, S10b, Table S1). A new Ru‐CO scattering is observed at 1.88±0.02 Å with a coordination number of 1.1±0.2 (Figure 2 c; Supporting Information, Table S1). The bond lengths are the same as the values reported for molecular mer(Cl)‐[Ru(bipy)(CO)Cl3] (Ru−N 2.08 Å, Ru−Cl 2.33 Å, and Ru−C 1.89 Å; Supporting Information, Table S5 complex D). [19] Similar bond lengths are also confirmed by the DFT calculations (Supporting Information, Figure S4b, Table S2). No obvious edge shift is observed in the XANES, suggesting the unchanged oxidation state of RuIII (Supporting Information, Figure S9a, S10a). We conclude that majority of the RuIII surface complex becomes mer(Cl)‐[bipy‐RuIII(CO)Cl3] via:
![Evolution from [bipy‐RuIIICl4]− to mer(Cl)‐[bipy‐RuIII(CO)Cl3]. a) Mass spectrometry (MS) showing the outlet gas profile with the corresponding temperature profile under 1 %CO and 5 %O2 flow, O2 is fed to monitor if there is any oxidation of CO to CO2. Blue CO, purple CO2, red O2, green HCl. b) In situ DRIFTS spectra, showing the formation of one terminal CO coordination at 2057 cm−1. c) Experimental and fitted EXAFS data, suggesting the [bipy‐RuIII(CO)Cl3] single‐site structures with corresponding DFT calculated models.](/dataresources/secured/content-1765837351133-55816efd-9798-4b19-954a-9e67aead3496/assets/ANIE-60-1212-g002.jpg)
Evolution from [bipy‐RuIIICl4]− to mer(Cl)‐[bipy‐RuIII(CO)Cl3]. a) Mass spectrometry (MS) showing the outlet gas profile with the corresponding temperature profile under 1 %CO and 5 %O2 flow, O2 is fed to monitor if there is any oxidation of CO to CO2. Blue CO, purple CO2, red O2, green HCl. b) In situ DRIFTS spectra, showing the formation of one terminal CO coordination at 2057 cm−1. c) Experimental and fitted EXAFS data, suggesting the [bipy‐RuIII(CO)Cl3] single‐site structures with corresponding DFT calculated models.
mer(Cl)‐[bipy‐RuIII(CO)Cl3] is relatively stable because any removal of the Cl− will result in the formation of unfavorable positively charged Ru complexes. With 100 mg of PPhen‐[bipy‐RuIIICl4]H, 1.25 mL of CO is consumed, corresponding to 5.6 wt % Ru loading on the initial PPhen‐[bipy‐RuIIICl4]H (Supporting Information, Figure S11). This observation is in good agreement with the 5.8 wt % Ru loading calculated from the Thermogravimetric analysis (TGA), proving the chemical stoichiometry of Equation (1).
Structural Evolution from mer(Cl)‐[bipy‐RuIII(CO)Cl3] to cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2]
A further increase in the temperature does not result in any significant changes in the online MS profile, in situ DRIFTS and XAFS spectra (Figure 2 a; Supporting Information, Figure S9). This shows that CO can only replace Cl−, while the reduction of Ru3+ requires H2. H2 is added to the feed in order to reduce Ru and remove another Cl− ligand. HCl is detected in the MS, suggesting the reaction between H2 and the Cl− ligand (Figure 3 a). DRIFTS and FTIR spectra show a second C≡O stretching vibration absorption at 1996 cm−1 when H2 is introduced (Figure 3 b; Supporting Information, Figure S12a). The far‐FTIR spectra shows only one type of Ru−Cl stretching at 331 cm−1 (Supporting Information, Figure S12b). These results match the FTIR spectrum of molecular cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] (Table S5, complex E) [20] but are different from cis(CO)‐cis(Cl)‐[RuII(bipy)(CO)2Cl2] (Supporting Information, Table S5, complex F, 2040 and 1980 cm−1). [20] From analysis of the EXAFS spectrum, the Ru−Cl coordination number is reduced to 2.2±0.3 with a slight increase of bond length to 2.39±0.01 Å, confirming the ligand exchange of Cl− via CO with the help of H2 (Figure 3 c; Supporting Information, Figure S10d and Table S1). As a result, the coordination number of Ru−CO at 1.88±0.01 Å increases to 1.9±0.3 (Supporting Information, Table S1). [21] DFT calculations are performed accordingly with cis(CO)‐trans(Cl)‐[Ru(bipy)(CO)2Cl2], showing similar Ru−Cl bond length (2.339 Å) and a slightly longer Ru−CO bond length (2.002 Å; Supporting Information, Figure S4c, Table S2).
![Structural evolution from mer(Cl)‐[bipy‐RuIII(CO)Cl3] to cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2]. a) Mass spectrometry showing the outlet gas profile with corresponding temperature profile: Blue CO, purple CO2, red O2, green HCl, orange H2. b) In situ DRIFTS spectra showing the formation of two terminal CO coordination at 2057 cm−1 and 1996 cm−1. c) Experimental and fitted EXAFS results, suggesting the [bipy‐RuII(CO)2Cl2] single‐site structures with corresponding DFT calculated models. XANES of [bipy‐RuIIICl4]− and cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2] at d) Ru L3‐edge and e) Cl K‐edge. f) Molecular orbital diagram of RuIII and RuII single sites.](/dataresources/secured/content-1765837351133-55816efd-9798-4b19-954a-9e67aead3496/assets/ANIE-60-1212-g003.jpg)
Structural evolution from mer(Cl)‐[bipy‐RuIII(CO)Cl3] to cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2]. a) Mass spectrometry showing the outlet gas profile with corresponding temperature profile: Blue CO, purple CO2, red O2, green HCl, orange H2. b) In situ DRIFTS spectra showing the formation of two terminal CO coordination at 2057 cm−1 and 1996 cm−1. c) Experimental and fitted EXAFS results, suggesting the [bipy‐RuII(CO)2Cl2] single‐site structures with corresponding DFT calculated models. XANES of [bipy‐RuIIICl4]− and cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2] at d) Ru L3‐edge and e) Cl K‐edge. f) Molecular orbital diagram of RuIII and RuII single sites.
Ru L‐edge and Cl K‐edge XAFS spectra were measured to determine the oxidation state of Ru. At Ru L3‐edge region, two absorption features A at 2838.0 eV and B at 2840.9 eV have been found for the initial [bipy‐RuIIICl4]− site (Figure 3 d; Supporting Information, Figure S13 and Table S6). According to previous research and in accordance with the dipole selection rule, [22] these two peaks are attributed to 2p3/2→4d t2g and 2p3/2→4d eg transitions (Figure 3 f), respectively. [23] This proves that Ru 4d t2g orbitals are not completely filled and are in the form of RuIII t2g 5eg 0, which is in good agreement with the Ru K‐edge XANES spectrum (Figure 1 c). For cis(CO)‐trans(Cl)‐[Ru(bipy)(CO)2Cl2] sites, feature A disappears, and a new feature C appears at 2843.0 eV. The absence of feature A suggests completely filled RuII 4d t2g 6 orbitals. Thus, the reduction from RuIII to RuII is confirmed. The additional feature C is assigned to Ru 2p3/2→CO π* transition.[ 22 , 24 ] The CO π* orbitals are formed above Ru 4d eg as a result of Ru−CO π backbonding. Similar electronic structures are also obtained from the Ru L2‐edge XAFS spectra (Supporting Information, Figures S13, S14). The Ru K‐edge spectra show no obvious changes, resulting from the dipole forbidden 1s→4d transition that prevents the detection of valence orbitals (Supporting Information, Figures S10c, S15, Table S7).
In Ru‐Clx complexes, d–p hybridization enables the transition from Cl 1s to Ru 4d, [25] thereby Cl K‐edge XAFS can also validate the Ru oxidation state. There is a strong pre‐edge absorption feature E at 2823.8 eV for both [bipy‐RuIIICl4]− and cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] sites in the Cl K‐edge XAFS, representing the Cl 1s→Ru 4d eg transition (Figure 3 e). The [bipy‐RuIIICl4]− site shows a feature D at 2820.8 eV, which is absent in cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] site. This feature originates from Cl 1s to unfulfilled RuIII 4d t2g. Therefore, the reduction is further verified via Cl K‐edge XAFS. It also suggests that the Cl is not an outer shell counter anion but is in a close coordination environment with Ru cation.[ 25b , 26 ] The energy differences between features A and B as well as D and E are 2.9 eV and 3.0 eV, respectively. Such ligand field splitting energy Δ0 at core hole state is between 23 400 to 24 200 cm−1, which corresponds well with the absorption at 352 nm in UV/Vis spectra (Supporting Information, Figure S16). Furthermore, XPS at the Ru 3d region also confirms the change of oxidation states, showing a decrease of the Ru 3d5/2 binding energy from 282.4 eV to 281.8 eV (Supporting Information, Figure S17 and Table S8). As such, we confirm that the reaction mechanism followed is:
Molecular O2 is constantly present in the system and an air stable cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] site is achieved.
CO Oxidation over RuII and RuIII Single Sites
Further increasing the temperature shows the release of CO2 from 503 K (Figure 3 a). This suggests that cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] is active for CO oxidation. We perform CO oxidation for both mer(Cl)‐[bipy‐RuIII(CO)Cl3] and cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] sites. The former shows no conversion, whereas the latter gives a light off at 388 K and 35 % conversion at 453 K, p CO=0.01 bar and weight hourly space velocity (WHSV) of 60,000 mL h−1 g−1 (Supporting Information, Figure S18). An activation energy (E a) of 90 kJ mol−1 is determined at the WHSV of 600 000 mL h−1 g−1. Based on the EXAFS study of the cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] site and its narrow full width at half maximum (FWHM) of CO absorption in FTIR spectra (Supporting Information, Figure S12a), we assume that all the RuII sites are identical and perform the same in CO oxidation. The corresponding TOFs are then calculated as 0.73×10−2 s−1, 1.77×10−2 s−1, and 3.89×10−2 s−1 at 462, 479 and 498 K, respectively. The TOF and E a are similar to the state‐of‐the‐art Cu and Pt single site over metal oxide support for CO oxidation (Supporting Information, Table S9). HAADF‐STEM images show the high density of Ru2+ cations well dispersed on the polymer framework after catalysis for cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] site (Supporting Information, Figure S19). There is no sign of aggregation or cluster formation, which is further proved via ex situ XRD studies (Supporting Information, Figure S19a–f). The RuII retains the cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] structure, as indicated in the XAFS (Supporting Information, Figure S19g–i) and FTIR (Supporting Information, Figure S12c) studies after catalysis. The PPhen‐bipy polymer remains unchanged, according to the post catalysis FTIR and scanning electron microscopy analysis (Supporting Information, Figure S12c and Figure S20). This is because of its high stability in air at elevated temperatures (Supporting Information, Figure S5c). The activity of cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] is stable during the 24 h on stream stability test (Supporting Information, Figure S21). The distinct differences in CO oxidation activity between mer(Cl)‐[bipy‐RuIII(CO)Cl3] single site and cis(CO)‐trans(Cl)‐[RuII(bipy)(CO)2Cl2] single site are resulted from their geometric and electronic properties. Geometrically, the former has three Cl− ligands, which are strong binding ligands in metal complexes (poisonous for metal active center) and leaves only one CO ligand to be exchanged during catalysis. In comparison, the latter offers two exchangeable CO ligands in cis configuration, enabling the local activation of CO within the Ru coordination environment. Electronically, the RuII is in a reduced form, which is reported to activate CO in water‐gas‐shift reaction. [27]
The role of the halogen ligand is further studied via [bipy‐RuIIIBr4]− single site, where Br− is a better leaving group than Cl− according to solution based coordination chemistry. The transition from [bipy‐RuIIIBr4]− to [bipy‐RuIII(CO)Br3] takes place at room temperature while the reduction from [bipy‐RuIII(CO)Br3] to [bipy‐RuII(CO)2Br2] requires 513 K (Supporting Information, Figure S22 and Table S2). This is 40 K higher than the temperature required to reduce the Cl coordinated Ru single sites. Without the presence of solvent, the bond breaking and formation here at the solid/gas interface are different.
General Synthetic Strategy
The homogeneity of the single sites is crucial to provide a key platform to investigate the structure–activity relationship. [28] Here in the PPhen‐bipy system, the bidentate nitrogen coordinative environment is well preserved. Therefore, the formation of surface [bipy‐MXn] site is predictable according to coordination chemistry. We further extend the scope of such system to twelve metal cations based on their positions in the periodic table and their applications in catalysis. They are 3d metals, including Fe3+, Co2+, Ni2+, Cu2+, and Zn2+, noble metals, including Rh3+, Pd2+, Ag+, Ir3+, Pt4+, and Au3+, and p‐block element Sn4+. This selection covers cations with valence electrons from d5 to d10 and ionic radius from 69 pm to 129 pm, which can form tetrahedral, square planar and octahedral geometries that lead to different catalytic behaviours. [bipy‐FeCl4]−, [bipy‐CoCl2], [bipy‐CuCl2], [bipy‐ZnCl2], [bipy‐RhCl4]−, [bipy‐PdCl2], and [bipy‐PtCl4]− are obtained with good metal dispersion over PPhen‐bipy, as determined in HAADF‐STEM and XAFS studies (Figure 4 a,b,d–h). XANES confirms that the oxidation states of metals are the same as the metal precursors (Supporting Information, Figure S23). Ni2+ forms [bipy‐Ni(H2O)4]2+ sites instead of [bipy‐NiCl4]2− (Figure 4 c). 4d and 5d single sites are easily recognized in the HAADF‐STEM compared to 3d metals, due to the higher Z‐contrast of these heavy metal cations. The BET equivalent surface area of PPhen‐[bipy‐RhCl4]− is 410 m2 g−1, similar to that of the PPhen‐[bipy‐RuIIICl4]− (Supporting Information, Table S4 and Figure S6). Ag, Sn, Ir, and Au form a majority of clusters instead of single sites (Figure 4 i–k), which is possibly due to the weak M−N/M−Cl coordination bond and the strong M−M or M−O bonds that promote cluster formation. Apart from metal cations, the selection of ligands is also controllable. [bipy‐RuCl4]−, [bipy‐RuBr4]−, [bipy‐CuCl2], and [bipy‐Cu(H2O)4]2+ are achieved accordingly (Supporting Information, Figure S24). The diverse selection of metal cations and ligands provides a single‐site palette with controllable geometric and electronic structures for a target catalytic reaction.
![HAADF‐STEM and EXAFS identifications of single site PPhen‐[bipy‐MXn] structure. Cs‐corrected HAADF‐STEM images show the single metal ions supported on PPhen‐bipy for a) Fe3+, b) Co2+, c) Ni2+, d) Cu2+, e) Zn2+, f) Rh3+, g) Pd2+, and h) Pt4+. Clusters and nanoparticles are observed in HAADF‐STEM images for PPhen‐bipy supported i) Ag+, j) Sn4+, k) Ir3+, and l) Au3+. Corresponded experimental and fitted EXAFS results are shown in a) [bipy‐FeCl4]−, b) [bipy‐CoCl2], c) [bipy‐Ni(H2O)4]2+, d) [bipy‐CuCl2], e) [bipy‐ZnCl2], f) [bipy‐RhCl4]−, g) [bipy‐PdCl2] and h) [bipy‐PtCl4], respectively. The coordination number and distance of M−Cl and M−N are listed in the Supporting Information, Table S3.](/dataresources/secured/content-1765837351133-55816efd-9798-4b19-954a-9e67aead3496/assets/ANIE-60-1212-g004.jpg)
HAADF‐STEM and EXAFS identifications of single site PPhen‐[bipy‐MXn] structure. Cs‐corrected HAADF‐STEM images show the single metal ions supported on PPhen‐bipy for a) Fe3+, b) Co2+, c) Ni2+, d) Cu2+, e) Zn2+, f) Rh3+, g) Pd2+, and h) Pt4+. Clusters and nanoparticles are observed in HAADF‐STEM images for PPhen‐bipy supported i) Ag+, j) Sn4+, k) Ir3+, and l) Au3+. Corresponded experimental and fitted EXAFS results are shown in a) [bipy‐FeCl4]−, b) [bipy‐CoCl2], c) [bipy‐Ni(H2O)4]2+, d) [bipy‐CuCl2], e) [bipy‐ZnCl2], f) [bipy‐RhCl4]−, g) [bipy‐PdCl2] and h) [bipy‐PtCl4], respectively. The coordination number and distance of M−Cl and M−N are listed in the Supporting Information, Table S3.
[bipy‐PtCl4]− and [bipy‐CuCl2] sites are studied in the CO oxidation under the same condition to that of the Ru single sites. Both show very limited CO conversion (Supporting Information, Figure S25), despite that Pt and Cu single sites over metal oxide support are active in this reaction (Supporting Information, Table S9). Such a difference between molecularly designed Ru and Cu/Pt sites can shed light on the future research.
Conclusion
We have designed an air‐stable cis(CO)‐trans(Cl)‐[bipy‐RuII(CO)2Cl2] single site over a PPhen‐bipy framework with molecular‐level accuracy. Compared to the completely inert RuIII site, the RuII site is active for CO oxidation owing to its unique electronic and geometric structure. Such a RuII single site can only be formed in the simultaneous RuIII to RuII reduction by H2 and the ligand exchange from Cl− to CO. With the help of a well‐defined pristine single site, such solid/gas interface coordination chemistry can be used to explain the surface bond breaking and formation at molecular level. The design method is generally applicable and is versatile for metal centers that are ubiquitous in catalysis, as supported by the synthesis of [bipy‐MXn] sites for another nine elements.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
The project is funded by EPSRC (EP/P02467X/1 and EP/S018204/2), Royal Society (RG160661, IES\R3\170097, IES\R1\191035, IES\R3\193038) and the Newton International Fellowship (NF170761). We acknowledge electron Physical Science Imaging Centre at Diamond Light Source (UK) for the STEM experiment (Proposal No. EM16966, EM17559, EM18909, EM19246, EM19318, EM20643, and EM20847). We acknowledge the B18 beamline of Diamond Light Source for XAFS experiment (Proposal No. SP17377). We thank the Diamond Light Source for the award of beamtime at B18 beamline as part of the UK Catalysis Hub Block Allocation Group (Proposal No. SP15151) and Energy Materials Block Allocation Group (Proposal No. SP14239). We acknowledge the BL6N1 beamline of Aichi Synchrotron Radiation Center (Japan) for the Ru L‐edge and Cl K‐edge XAFS measurements (Proposal No. 20190464). We acknowledge Helmholz‐Zentrum Berlin for the beamtime at ISISS beamline in BESSY II (Proposal No. 191‐08389 ST). We thank the UK Catalysis Hub for resources and support provided via membership of the UK Catalysis Hub Consortium and funding by EPSRC (grants EP/ K014706/2, EP/K014668/1, EP/K014854/1, EP/K014714/1 and EP/M013219/1). We thank the Research Complex for access and support to the facilities and equipment.
References
1
2
2b
2c
2d
3
3a
3c
3e
4
4a
5
6
7
7b
8
9
10
10a
10b
11
12
13
13
15
16
17
18
19
19a
19b
20
20a
20b
21
21a
21b
21c
21d
22
23
23a
23b
23c
23d
24
25
25a
25b
27
27a
27b
28
28a
28b
28b
28c
28d